FDA Continues Push to Improve Food Labeling Practices in the United States

In September 2022, former President Biden convened the White House Conference on Hunger, Nutrition, and Health, during which the White House introduced its National Strategy on Nutrition and Health (National Strategy). The National Strategy called for creating more accessible food labeling practices to empower consumers to make healthier choices, among other laudable public health-focused goals. Prior to the January 2025 transition from the Biden to the Trump administration, the Food and Drug Administration (FDA) took concrete steps to address this particular National Strategy priority through both formal rulemaking and informal guidance. This blog post summarizes FDA’s actions at the end of the Biden administration intended to modernize food labeling practices and move them forward in today’s more consumer-focused marketplace.
Proposed Rule for Front-of-Package Nutrition Labeling
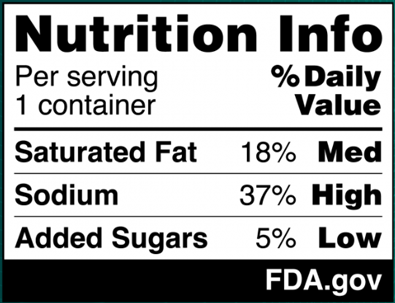
In the National Strategy, the development of front-of-package (FOP) labeling schemes was discussed as one way to promote equitable access to nutrition information and healthier choices. On January 16, 2025, FDA published in the Federal Register a proposed rule that would require a front-of-package nutrition label on packaged foods (Proposed Rule). The Proposed Rule would require manufacturers to add a “Nutrition Info” box on the principal display panel of each packaged food product, which would list the Daily Value (DV) percentage of saturated fat, sodium, and added sugars in a serving of that food. The DV percentage would list how much of the nutrient in a serving contributes to a person’s total daily diet. In addition, each of those nutrients would include corresponding “interpretative information” that would signal to consumers whether the food product contains a low, medium, or high amount of those nutrients. An example of the proposed FOP nutrition information graphic is below. And although the Proposed Rule would not require it, manufacturers could voluntarily include a calorie count on the front of the food package, per existing FDA regulations.
The Proposed Rule does deviate from certain suggestions made in the National Strategy, which advocated for FOP “star ratings” and “traffic light schemes” to promote equitable access to nutrition information. Specifically, the National Strategy considered how to best help consumers with lower nutrition literacy more readily identify foods that comprise a healthy diet. Instead of a front-of-packaging labeling system that would rely on imagery, however, FDA’s proposal opted for written information about the nutrients contained in the food. Both the preamble to the Proposed Rule and FDA’s press release announcing its publication explain that in focus groups conducted in 2022, participants reported confusion over the traffic light system in particular (e.g., when a food contained both nutrients that should be limited but also nutrients for which higher consumption is recommended) and that “the black and white Nutrition Info scheme with the percent [DV] performed best in helping consumers identify healthier food options.”
It will be interesting to see whether comments to the Proposed Rule will remark on FDA’s choice of the written “Nutrition Info” box versus a FOP labeling system that would be more reliant on imagery. FDA is accepting comments on the Proposed Rule until May 16, 2025 (Docket FDA-2024-N-2910). As currently envisioned, if the proposal for FOP nutrition information is adopted, most food product manufacturers would have three years from the effective date to bring labels into compliance (smaller manufacturers would be given four years).
As a result of President Trump’s administrative freeze and new executive orders governing the work of regulatory agencies such as FDA, the fate of this Proposed Rule is currently uncertain. However, newly confirmed Health and Human Services (HHS) Secretary Robert F. Kennedy Jr. has articulated that his agenda is to “make America healthy again” (MAHA) and the presidential MAHA Commission was recently established to begin informing the Administration’s work on Mr. Kennedy and President Trump’s priorities in this space. Although Mr. Kennedy did not address food labeling during his Senate confirmation hearings and the executive order creating the MAHA Commission does not speak directly to food labeling or nutrition information accessibility for consumers, interested stakeholders should monitor the upcoming work of the Commission – including whether any opportunities for public comments may be made available – as well as its future “Make Our Children Healthy Again Strategy” that is due in approximately six months. Further, under a deregulatory executive order signed on January 31, 2025, President Trump has directed agencies to eliminate 10 “regulations” for each new regulation to be promulgated, with the term “regulation” expansively defined to include memoranda, guidance documents, policy statements, and interagency agreements. This “one-in, 10-out” order may make the prospect of an FOP nutrition labeling final rule less likely, at least for the foreseeable future.
Final Rule for Use of The Term “Healthy” on Food Labeling
Another recent FDA action related to food labeling was the agency’s finalization of a proposed rule from 2022 that involved a lengthy public consultation and information collection process (see our prior coverage here). On December 27, 2024, FDA published in the Federal Register its Final Rule regarding the use of the term “healthy” in food labeling. The Final Rule updates the definition established 30 years ago for the nutrient content claim “healthy” to be used in food labeling. In President Biden’s National Strategy, one highlighted priority was ensuring that food packages bearing this claim align with current nutrition science and the Dietary Guidelines for Americans (Dietary Guidelines). To advance this goal, FDA was charged with updating the standards for when a company can use the “healthy” claim on its products (work on which was already ongoing at the agency), creating a symbol that can be used to reflect that the food is “healthy, and developing guidance on the use of Dietary Guideline statements on food labels.
The original regulatory definition of “healthy” (codified at 21 C.F.R. § 101.65(d)) sets limits on total fat, saturated fat, cholesterol, and sodium content should a food be labeled as healthy, and requires that the food contain at least 10% of the DV for vitamin A, vitamin C, calcium, iron, protein, and fiber. Under the Final Rule, total fat and dietary cholesterol are no longer factors to be considered when evaluating whether a food is eligible for this particular nutrient content claim. Instead, the agency has established limits on saturated fat, sodium, and added sugars in accordance with the Dietary Guidelines. Additionally, rather than focusing on vitamin A, vitamin C, calcium, iron, protein, and fiber, the Final Rule requires that the food product contain a certain amount of food from at least one of the food groups or subgroups recommended by the Dietary Guidelines, such as fruit, vegetables, grains, dairy, and proteins.
Perhaps most notably, the prior regulatory scheme allowed for foods that were high in added sugars, such as yogurts, breakfast cereals, and fruit snacks, to technically qualify as “healthy” despite not aligning with the definition of “nutrient-dense” foods from the Dietary Guidelines, which specifically applies to certain foods “when prepared with no or little added sugars, saturated fat, and sodium.” Consistent with generally accepted nutritional best practices, the National Strategy also promoted lowering the sodium content in food and decreasing the consumption of added sugars –shared goals of new HHS Secretary Kennedy and the broader MAHA agenda.
The Final Rule does not establish a “healthy” symbol that can be used on food packaging, but FDA has indicated that this symbol may also be on the horizon. In its press release announcing the Final Rule, FDA noted that it is “continuing to develop” this symbol, adding that such a symbol would further FDA’s goal of helping consumers more easily identify healthier food products.
The Final Rule’s effective date (which as of publication of this blog post, has not been changed by the Trump Administration) is February 25, 2025, and the compliance date for manufacturers is February 25, 2028 – three years after the new regulatory definition becomes effective.
Draft Guidance for Industry: Labeling of Plant-Based Alternatives to Animal-Derived Foods
Finally, while not specifically called out in the National Strategy, FDA has been working for several years to develop labeling recommendations for plant-based foods that are being developed and marketed as alternatives to conventional animal products. On January 7, 2025, FDA released the Draft Guidance for the Labeling of Plant-Based Alternatives to Animal Derived Foods (Draft Guidance), in response to the growing demand for plant-based food alternatives in the United States. According to the Plant-Based Food Association, 70% of Americans are consuming plant-based foods. The scope of the newly released guidance encompasses alternatives to poultry, meat, seafood, and dairy products that fall under FDA’s jurisdiction. It expressly excludes plant-based milk alternatives, as separate guidance on that subject was released in February 2023.
The Draft Guidance notes that rather than simply identifying a product as a “plant-based” alternative food, the specific plant source should be disclosed on the food product’s label. This would enable consumers to make more informed choices about purchasing plant-based alternatives. For example, rather than labeling a plant-based cheese solely as such, the cheese’s label should more clearly disclose “soy-based cheese” to reflect its primary ingredients. The Draft Guidance also recommends that if a plant-based alternative food is derived from several different plant sources, the primary plant sources should be identified in the food’s name. The agency provides the examples of “Black Bean Mushroom Veggie Patties” and “Chia and Flax Seed Egg-less Scramble” to illustrate this concept. For labeling purposes, FDA also recommends companies avoid exclusively naming products with “vegan,” “meat-free,” or “animal-free.”
Public comments on the Draft Guidance should be submitted by May 7, 2025 (Docket FDA-2022-D-1102).
Conclusion
One primary goal of the National Strategy was to empower Americans to make healthier, informed choices about their nutrition and food consumption. In the United States, diet-related diseases, such as hypertension, obesity, and diabetes, are on the rise. Under the Biden administration and the leadership of former Commissioner Dr. Robert Califf, FDA sought to fight these alarming trends and to improve public health by increasing access to nutritional information and promoting transparency in food labeling.
Further, while the Proposed Rule, Final Rule, and Draft Guidance all focus on labeling packaged food products that can be purchased in stores, it will be interesting to see how these initiatives influence FDA’s recommendations for food labeling practices in online grocery shopping. On April 24, 2023, FDA published the notice Food Labeling in Online Grocery Shopping; Request for Information (Docket No. FDA-2023-N-0624-0002), which received 31 electronically submitted comments from various stakeholders, including grocer organizations, food scientists, and individual consumers. Indeed, the December 2024 press release for the Final Rule noted that FDA “has already entered into a partnership with Instacart to make it even easier for consumers to find products with the ‘healthy’ claim through online grocery shopping filters and a virtual storefront.” In the wake of the agency actions summarized in this post and the Instacart partnership, we wonder if FDA will move in the future to provide manufacturers and retailers with definitive guidance on online food labeling practices. We will be watching to see how FDA, as well as the work of the MAHA Commission and HHS Secretary Kennedy, may continue to improve food labeling practices in the future.
Navigating the EPR Laws: What Alcohol Beverage Producers Need to Know
Extended producer responsibility (EPR) laws are relatively new – the first were signed into law in 2021 and 2022 – and are aimed at encouraging producers to package goods in a more environmentally conscientious manner and providing much needed revenue for in-state recyclers overwhelmed by the incoming volumes of recyclable material. In essence, EPR laws require producers whose products reach in-state consumers to register with a producer responsibility organization (PRO) or stewardship organization (SO), report the amount of material that enters the state, and pay fees for that material.
As detailed in this article, how and if these new laws apply to alcohol beverage suppliers is nuanced given both the developing nature of EPR laws as well as the interplay with preexisting container deposit or bottle bill laws.
Who and What Is Covered by the New EPR Laws?
As of the writing of this article, five states have passed EPR laws and 10 others have introduced bills during their most recent legislative sessions, including Connecticut, Hawaii, Nebraska, and New York. EPR programs are still evolving as the various regulatory processes continue; however, once passed by a state, the new rules typically require the state to designate a PRO/SO to administer the producer requirements and aid producers and state agencies with the necessary reporting and payment requirements. For those states that have passed EPR laws, the nonprofit organization Circular Action Alliance is the PRO/SO that has been selected to oversee the process.
Each EPR law has its own definition of “producer” (i.e., those required to report) and of “covered material” (i.e., the materials that must be reported). There are several specifics within each state but, generally, the producer is the entity who actually produces the subject goods or the owner of the brand that is contracting to have the item produced. Speaking broadly, the covered material contemplated by these laws includes the cardboard boxes and glass or aluminum containers that protect and contain the items purchased from retailers. In this regard, EPR laws apply to alcoholic beverages, non-alcoholic beverages, and nearly all other types of consumer goods.
However, exemptions exist for small producers and certain materials. The specifics as to when a producer is exempt from registering and making EPR payments vary by state but, in many instances, those producers shipping small amounts into a certain state will be exempt. For example, Minnesota exempts producers responsible for less than one metric ton of covered material or $2,000,000 in global gross revenue. However, it is important to remember that even if a small producer is, given its status, exempt from registering with a PRO/SO or paying the related EPR fees, it will likely still have reporting requirements to substantiate its claims of being exempt.
How Do These Laws Apply and Interface with Alcohol Beverage Laws?
In the world of beverage alcohol, there are numerous laws already in place, many of which have been in force for decades, that are aimed at sustainability and designed to fund and encourage recycling. These are referred to as beverage container deposit/recycling deposit requirements or “bottle bills.” These laws generally require subject beverages to have, listed on their labels, their deposit amount or to otherwise identify the bottle as one that is subject to deposit requirements. They also require a small deposit to be paid by the retailer and/or by the consumer for the purchase of subject beverages, only refunding the deposit to the consumer when the bottle is returned and requiring a reimbursement and handling fee to be paid by distributors and/or manufacturers for the processing/recycling of the containers.
The exact recycling model, deposit collection, and payment process varies by state. For instance, California does not require the amount of the deposit to be listed on the bottles and requires the distributor and the manufacturer/importer to remit payments, but it does not require the retailer to submit payments, as the retailers are generally acting as the redemption centers for consumers.
Fortunately, many of the EPR laws specifically exempt items subject to beverage container deposit requirements. However, the interplay between the older beverage container deposit requirements and the newer EPR laws are not always straightforward. For example, in Oregon, “beverage containers” as defined by the recycling deposit regulations are excluded from the definition of “covered products” (i.e., those subject to EPR requirements). But only “water or flavored water; beer or another malt beverage; mineral water, soda water, or a similar carbonated soft drink; kombucha; or hard seltzer” are subject to deposit regulation, not wine or spirits. This means, for a producer that is selling wine, spirits, and malt beverages into Oregon, while they would be able to exclude the weight of the cases for the malt beverages (malt cans/bottles would be exempt), they would still have to calculate the weight of the bottles and cases for the wine and spirits when reporting.
In short, given the developing nature of these laws, coupled with the nuances of their interplay with other alcohol beverage-specific requirements, there is no one-size-fits-all analysis as to how and when these laws apply to alcohol beverage producers. There are some instances where an alcohol-specific exemption may apply and other states where no such exemption exists. To ensure compliance in this rapidly evolving landscape, each company’s business structure must be examined carefully to determine whether it, or the products it ships, are subject to EPR laws, bottle bills, or both.
McDermott+ Check-Up: February 21, 2025
THIS WEEK’S DOSE
Senate Passes Budget Resolution. The “skinny” bill was put on the Senate floor shortly after President Trump expressed support for the House’s version.
Administration’s Federal Workforce Cuts Hit HHS. Thousands of employees were let go across the divisions of the US Department of Health and Human Services (HHS).
President Trump Issues Several EOs. The executive orders (EOs) relate to in vitro fertilization, COVID-19 vaccine mandates, independent agencies, deregulation, and the federal workforce.
Legal Challenges Continue to Block Gender-Affirming Care EO. A federal judge issued a second 14-day stay as the court considers the legality of the order.
CONGRESS
Senate Passes Budget Resolution. Last week, the Senate and House Budget Committees each passed separate, and very different, budget resolutions as their first steps toward negotiating a unified budget resolution that must pass both bodies in order for work to proceed on reconciliation. These resolutions reflected each chamber’s preferred approach. The Senate is moving a two-part reconciliation strategy by advancing a “skinny” resolution that only addresses immigration, energy, and defense priorities (but which still may utilize healthcare as a pay for). The Senate would act later to advance a separate resolution to extend the 2017 tax cuts. The Senate’s goal is to provide President Trump with a quick win, then take the additional time members think will be necessary to pass a reconciliation package tackling tax cuts. In contrast, the House is proceeding with a budget resolution that includes tax cuts and a minimum of $1.5 trillion in spending reductions. The House approach clearly puts healthcare on the table for significant cuts. Medicaid is a particular focus given that the resolution would require the House Energy & Commerce Committee to come up with $880 billion in savings.
While the House was in recess this week, Senate Majority Leader Thune (R-SD) scheduled a vote on the recently advanced Senate budget resolution. Then, on February 19, President Trump endorsed the House’s one-big-bill approach. This Senate still moved forward with the scheduled vote, passing the resolution 52 – 48 and indicated that doing so will provide a backstop if House efforts fail. Senator Paul (R-KY) was the only Republican to vote no.
House Republican leaders plan to bring their budget resolution to the House floor as soon as next week, but the timing is uncertain as several Republican members of Congress have expressed hesitation about supporting it. Some are Republicans in swing districts who are concerned about the magnitude of Medicaid cuts. Others are members who oppose voting to increase the debt limit, which is also included in the budget resolution.
ADMINISTRATION
Administration’s Federal Workforce Cuts Hit HHS. Over the weekend, the Trump administration reduced HHS’s workforce by several thousand employees across several agencies, including the US Food & Drug Administration, the Centers for Medicare & Medicaid Services (CMS), the Centers for Disease Control and Prevention, and the National Institutes of Health. Many who were let go had probationary status (meaning they were hired or promoted less than a year ago) or temporary status (which could include employees who have spent years in their role). The laid-off employees had worked on a variety of issues, such as Medicare and Medicaid quality initiatives, medical device approvals, public health preparedness, and artificial intelligence. At this time, there is no transparency as to the positions eliminated or even the overall counts. Per a recent EO, the agencies could be restricted from adding staff, as the EO permits hiring of no more than one employee for every four employees that depart.
President Trump Issues Several EOs. The administration continues to highlight and implement its agenda through EOs. Relevant EOs issued this week include the following:
IVF. This EO directs the assistant to the president for domestic policy to submit a list of policy recommendations to protect in vitro fertilization (IVF) access and reduce the out-of-pocket and health plan costs for the treatment. The fact sheet can be found here. Like many other EOs, additional steps would need to be taken before any changes occurred.
COVID-19 Vaccine Mandates. This EO mandates the withholding of federal funds from educational entities that require students to receive a COVID-19 vaccination to attend in-person education programs. It requires the secretaries of education and HHS to issue guidelines for compliance, a report on noncompliant entities, and a planned process for each agency’s implementation. The fact sheet can be found here. It is unclear how much practical impact this EO may have, because most of these directives have ceased to be enforced.
Independent Agencies. This EO requires independent agencies, including the Federal Trade Commission, to submit proposed regulations to the Office of Information and Regulatory Affairs before publication in the Federal Register. The EO directs the Office of Management and Budget (OMB) to establish performance standards and management objectives for independent agencies and to review independent agency actions for consistency with the president’s priorities. The EO also states that only the president and attorney general can provide interpretations of law for the executive branch.
Deregulation. This EO directs agency heads to work in coordination with Department of Government Efficiency team leads and OMB to review all regulations subject to their jurisdiction for consistency with law and administration policy. Within 60 days, agencies must submit to OMB a list of certain regulations, including those that are unconstitutional, are not authorized by statutory authority, and impose undue burdens on small businesses. The EO states that agencies should deprioritize actions that enforce regulations that go beyond the powers vested by the Constitution and should ensure that enforcement actions are compliant with law and administration policy. The EO also directs OMB to issue implementation guidance.
Federal Workforce. This EO requires HHS to terminate the secretary’s advisory committee on long COVID-19, and CMS to terminate the health equity advisory committee. It also directs non-statutory components and functions of certain foreign affairs governmental entities to be eliminated, as allowed under applicable law, and directs such entities to submit a report stating whether components of the entity are statutorily required.
COURTS
Legal Challenges Continue to Block Gender-Affirming Care EO. Lawsuits continue to be filed against actions taken by the Trump administration, including EOs and other administrative announcements. This includes a lawsuit filed by the attorneys general of Washington, Oregon, Colorado, and Minnesota, along with three doctors who provide gender-affirming care to youth. On February 14, a federal judge issued a two-week temporary restraining order that blocks the withholding of funds to healthcare entities that provide gender-affirming care to patients under 19. This is the second judge to take action on this EO. On February 13, another judge issued a two-week temporary restraining order blocking enforcement of the EO.
QUICK HITS
CBO Publishes Explainer on Scoring. The document explains how the Congressional Budget Office (CBO) prepares cost estimates for legislation. This process is top of mind for stakeholders as the budget reconciliation process (which is expected to include healthcare-related budgetary offsets) continues.
NEXT WEEK’S DIAGNOSIS
Congress will be in session next week, with the House potentially voting on its budget resolution. The Senate will continue work to confirm President Trump’s nominees, including a nomination hearing for Dan Bishop as deputy director of OMB. Health-related hearings include:
A House Energy & Commerce Health Subcommittee hearing on pharmacy benefit managers.
A House Veterans’ Affairs Committee hearing on electronic health record modernization.
A House Oversight and Government Reform Committee hearing on the US Government Accountability Office’s 2025 high-risk list.
A Senate Special Committee on Aging hearing on the opioid epidemic.
We expect the administration to continue taking executive actions related to healthcare.
Make Food “Healthy” Again: FDA’s Resolution for a Healthier 2025
The U.S. Food and Drug Administration (FDA) began 2025 with a resolution to make food “healthy” again by announcing a trio of new final and proposed rules that are intended to make it easier for consumers to identify healthy food choices. These rules – which include a new ban on the use of red dye No. 3, a revised definition of what “healthy” claims can be made about foods, and new proposed requirements for nutritional labeling on the front of food packaging – have implications for many stakeholders in the food industry.
Although these healthy food initiatives were initiated under the Biden Administration, they directly align with the new Trump Administration’s agenda for the U.S. Department of Health and Human Services (HHS). The recently confirmed Secretary of HHS under the second Trump administration, Robert F. Kennedy Jr., has campaigned on limiting the use of food dyes and taking aim at highly processed “junk foods” that contribute to obesity. Therefore, we expect that these new rules will be here to stay, despite the new administration’s recent freeze on new proposed rules and recommended postponement of new final rules (which is currently pending court review). Regulated stakeholders should take steps now to prepare for compliance.
Revocation of Use of Red Dye No. 3 in Food and Ingestible Drugs
On January 16, 2025, FDA announced that it amended its color additive regulations to revoke the authorization for the use of FD&C Red No. 3 (commonly known as “red dye No. 3”) as a color additive in food and drugs (90 Fed. Reg. 4628 (Jan. 16, 2025)). Therefore, effective January 15, 2027, red dye No. 3 will no longer be permitted for use in food products, such as candy, cakes, cupcakes, cookies, frozen desserts, frostings, and icings, and all certificates for the use of red dye No. 3 in such products will cease to be effective. Red dye No. 3 will be banned from use as a color additive in ingestible drugs, effective January 18, 2027. Any food and ingestible drug products (including imports) containing red dye No. 3 without effective certification will be considered adulterated.
Notably, although other countries still permit certain uses of red dye No. 3 (sometimes called “erythrosine” abroad), foreign manufactured food products imported into the U.S. will also need to comply with this new ban. Though FDA has not yet signaled whether the implementation of this new rule will be delayed further, food industry members should take steps now to reformulate any products containing red dye No. 3.
FDA’s action on red dye No. 3 is not surprising to some extent, considering that the agency revoked use of the dye in cosmetics in 1990, and its use is already prohibited in food in other countries. What remains to be seen is whether FDA will continue down the regulatory path it has begun. Petitions are presently before the agency to ban other chemicals in food use, such as PFAS, BPA, TCE, and titanium dioxide—some of which have been pending for years. Further, prior to the change in administrations, FDA announced that it is developing a “systemic process” for conducting post-market assessments of chemicals in food, including GRAS ingredients, color additives, food contact substances, and potential contaminants, and established a docket for public comment. By the time the comment period closed, more than 34,000 comments had been submitted to FDA, signaling significant interest in this issue. Given the new HHS Secretary’s stated priorities, it would not surprise us if FDA continues its rulemaking efforts in this space.
Updated Requirements for “Healthy” Claims
In recent years, FDA has expressed concern over the growing prevalence of preventable chronic diseases and health conditions associated with unhealthy diet choices. Because FDA’s research demonstrates that U.S. consumers choose foods based on information readily (and easily) available to them, FDA has focused its attention on revising its food labeling regulations to provide consumers with additional information about a food’s nutritional benefits (or lack thereof). The result is the revised final rule for “healthy” claims and a proposed rule mandating front-of-pack nutrition disclosures, as discussed below.
The preambles to these rulemaking efforts demonstrate that FDA’s thinking has been heavily influenced by the Dietary Guidelines for Americans, 2020-2025 (the “Dietary Guidelines”). Under the National Nutrition Monitoring and Related Research Act of 1990, the U.S. Department of Agriculture (USDA) and HHS must publish the Dietary Guidelines at least once every five years, based on the current state of scientific and medical knowledge. The current Dietary Guidelines deemphasize the importance of individual nutrients or food groups in isolation in favor of a more holistic approach that focuses on dietary patterns during different life stages. A healthy dietary pattern, according to the Dietary Guidelines, emphasizes nutrient dense foods across all food groups, while staying within calorie limits and limiting sugars, saturated fats, and sodium.
FDA regulations have included parameters on “healthy” claims since 1994, but the requirements have not significantly changed since that time, even though nutrition science has evolved. The parameters established under the original rule were fairly rigid, and included specific limits on total fat, saturated fat, cholesterol, and sodium, as well as minimum amounts of nutrients whose consumption was encouraged (e.g., vitamin A, iron, protein). Under the 1994 rule, a food had to meet all the limits of the “discouraged” nutrients and contain the minimum amount of at least one of the “encouraged” nutrients to bear a “healthy” claim. That had the effect of excluding certain “nutrient dense” foods often viewed by consumers as healthy, such as salmon (due to its fat content).
The new Final Rule (89 Fed. Reg. 106,064 (Dec. 27, 2024)) proposes a more flexible approach consistent with the revised Dietary Guidelines. Current nutrition science emphasizes nutrient-dense foods – such as fruits, vegetables, and whole grains – as part of a healthy dietary pattern. Nutrient-dense foods and beverages are defined as those that provide vitamins, minerals, and other health promoting nutrients but also have little or no added sugars, saturated fats, or sodium. Accordingly, foods that meet the requirements for “healthy”, as defined in the new rule, are foods that, because of their overall nutrition profiles, can be the “foundation” or “building blocks” of an overall healthy dietary pattern recommended by the Dietary Guidelines.
Under the rule, a “healthy” food claim “[s]uggests that a food, because of its nutrient content, may be useful in maintaining healthy dietary practices, where there is also implied or explicit information about the nutrition content of the food (e.g., “healthy”).” See 89 Fed. Reg. 106,064, 106161. In general, to meet the new parameters for “healthy” food claims, including claims that foods are “healthful” or “healthier”, food products (including individual foods, mixed products, main dishes, and meals) must (1) for example, contain a certain amount of food (referred to as a “food group equivalent”) from at least one of the food groups or subgroups recommended by the Dietary Guidelines for Americans (e.g., vegetables, fruits, whole grains, fat-free or low-fat dairy, lean meat, seafood, eggs, beans, peas, lentils, nuts, or seeds) and (2) meet specific limits for added sugars, saturated fats, and sodium (based on a percentage of the Daily Value (DV) for these nutrients). Under the new criteria, some categories of foods – vegetables, fruits, seafood, lentils, nuts and seeds, among others – automatically qualify for the “healthy” claim due to their nutrient density, so long as they do not contain any additional ingredients other than water. And some foods that did not qualify for a “healthy” claim under the old rule due to their fat content now do, including avocados, salmon, and olive oil. Conversely, some food products that qualified as “healthy” under the old rule now do not, including fortified white bread, highly sweetened yogurts, and highly sweetened cereal.
The new rule also contains a substantiation requirement. Manufacturers of foods for which “healthy” claims are made must make and keep written records substantiating the “healthy” claims in accordance with the new requirements, except where nutritional food labeling makes clear that the requirements are met.
The food industry may begin voluntarily complying with the new rule on or after February 25, 2025, and must comply by February 25, 2028. Therefore, the food industry should take steps now to review any “healthy” claims made for their products, assess whether those claims should be changed in light of the new final rule, and document the substantiation for their claims.
We add that health claims on food labeling are heavily policed by consumer class action attorneys under consumer fraud laws (particularly in California and New York), and that compliance with FDA’s requirements for “healthy” claims will not necessarily preempt these lawsuits, if a court determines that other aspects of the label or packaging renders the claim misleading on the whole. Therefore, food manufacturers should work closely with experienced counsel in formulating “healthy” claims and ensuring that these claims are properly substantiated.
Nutrition Labeling
On January 16, 2025, FDA published a proposed rule (90 Fed. Reg. 5426 (Jan. 16, 2025)) (the “Proposed Rule”), which, if finalized, would require a front-of-package nutrition label called a “Nutrition Info box” on most packaged foods to assist consumers in more easily identifying healthy foods. This new label would require manufacturers to address the relative amounts of saturated fat, sodium, and added sugars in a serving of the food, and identify whether those amounts are low, medium, or high. FDA’s proposed format for the new label appears below:
The ranges that FDA proposes for each category are as follows:
Low: 5% daily value (DV) or less
Medium: 6% to 19% DV
High: 20% DV or more
In proposing these DV ranges, FDA considered the regulatory history of the percent DV; the agency’s commitment to helping consumers understand the percent DV concept in the context of a person’s daily diet; and the agency’s existing regulatory definitions for nutrient content claims (including definitions established for “low” and “high” claims), among other factors. FDA believes that the ranges it proposes for the interpretive descriptions – and in particular, its designation of 5% DV or less as “low” and 20% DV or more as “high” – align with its longstanding regulatory approach.[1] However, FDA invites comment on its conclusions and analysis.
Calorie disclosures
In the Preamble to the Proposed Rule, FDA acknowledged that some manufacturers already voluntarily include a calorie statement on the front of the food packaging, in accordance with existing regulations. FDA invites comment from industry stakeholders on the inclusion of a mandatory or voluntary statement of calories in the proposed Nutrition Info box, as well as suggestions as to how FDA could consider including quantitative calorie information (e.g., “low”, “medium”, “high”) in the box (including any new data or other information on which FDA could base this interpretation).
Anticipated costs
One notable aspect of the Proposed Rule is its anticipated cost to industry, which FDA analyzed as part of the rulemaking effort. FDA quantified the estimated costs of relabeling to the packaged food industry as a whole to range between $66 million and $154 million per year over a ten-year time horizon. Further, although product reformulation is not a requirement or a stated goal of the Proposed Rule, FDA recognizes that the rule may result in voluntary reformulation efforts by some food producers. FDA estimates that the annualized costs of reformulation would range from $125 million to $377 million over a ten-year time horizon. FDA recognizes the possibility that some of these relabeling/reformulation costs may be passed along to consumers (at least in part).
If finalized, businesses with $10 million or more in annual food sales will be expected to comply with these requirements within three years of the final rule’s effective date. Businesses with less than $10 million in annual food sales will be expected to comply with the requirements within four years of the final rule’s effective date. FDA is accepting comments on this new proposed rule by May 16, 2025. Food industry stakeholders should consider submitting comments to this proposed rule. Foley’s Food and Beverage Industry Team members have extensive experience assisting regulated stakeholders in preparing comments on FDA rulemaking. Any of the authors would be happy to provide additional information.
Regulatory Freeze
As is standard practice for an incoming administration, one of President Trump’s first Executive Orders (“EO”) places a freeze on all pending regulations proposed in the last days of the Biden administration. The EO encourages a 60-day review of any such proposed rules or guidance, with further consultation by the Director of the Office of Management and Budget (OMB) as-needed for “for those rules that raise substantial questions of fact, law, or policy.”
Next Steps
Though there is some uncertainty about timing, overall, industry should plan for the possibility that these rules and orders will take effect. Therefore, food industry stakeholders should act now to:
Begin planning on reformulation of products with red dye No. 3;
Review and assess what “healthy” food claims are made about their products, and document their support for those claims in accordance with the new “healthy” framework; and
Consider submitting comments to the new proposed rule requiring front-of-package nutrition information labeling.
[1] FDA does not have a regulation that establishes a “medium” nutrient content claim for any nutrient. In the Preamble to the Proposed Rule, FDA effectively takes a common-sense approach to defining that term. See 90 Fed. Reg. 5426, 5445 (“The adjective ‘medium’ is defined as, for example, ‘being in the middle between an upper and lower amount, size, degree, or value . . . and ‘intermediate in quantity, quality, position, size, or degree’ . . . . The common meaning of the adjective ‘medium’, then, aligns with the meaning of the interpretive . . . we are proposing.”). Which is just to say that the “low” and “high” goalposts are really what matter for purposes of comment on the Proposed Rule.
China on the Move: Lessons from China’s 2024 National Negotiation of Drug Prices
China’s share of the global drug development pipeline grew from 3% in 2013 to 28% in 2023, positioning China as the second-largest region for clinical trials after the United States. Additionally, the proportion of drugs launched first in China increased from 9% in 2017 to 29% in 2023, placing China just behind the United States in terms of first-in-class launches. This trend highlights the contributions of domestic companies, whose pipelines are replenishing the global pharmaceutical landscape. As a result, NextPharma estimates that the combined value of China’s licensing-out deals reached around $46 billion in 2024, up from $38 billion in 2023 and $28 billion in 2022.
On the demand side, from 2019 to the first quarter of 2023, the National Healthcare Security Administration (NHSA) allocated 60% of savings from generic drug procurement to innovative drugs listed on the National Reimbursement Drug List (NRDL). This shift mirrors trends in developed markets where patented drugs dominate sales. By 2023, innovative drugs accounted for 15.1% of hospital drug expenditures in sample hospitals, up from less than 10% in 2018. However, affordability remains a challenge, which is significant as China continues to push for increased access to cutting-edge therapies.
The 2024 NRDL negotiations, which concluded in November 2024, offer insights into how China is addressing these affordability concerns while seeking to ensure access to innovative medicines. This GT Advisory explores five key takeaways from the 2024 NRDL negotiations and their potential implications for the future of innovative drug pricing and reimbursement in China.
A Contradiction Between NRDL Outcomes and the Growing Influence of Chinese Companies in Global Innovation
Support for First-in-Class and Innovative Drugs
BMI Fund Sustainability
A Continuous Dilemma for Multinational Companies (MNCs)
Reimbursement Coverage Expansion: Category C and Commercial Health Insurance
Continue reading the full GT Advisory.
It’s The Hope That Kills You: My Experience with Alabama’s Medical Cannabis Experiment
If you haven’t seen Ted Lasso, you need to do that immediately (after reading this). In the final episode of the first season, Ted Lasso – an American football coach hired, for reasons that don’t matter here, to coach an English football team – who is played exquisitely by an endearing Jason Sudeikis – notes that he has heard the expression “it’s the hope that kills you” when describing the trials and tribulations of English football. I will leave my thoughts on the merits of relegation and promotion in American sports for another forum.
It’s a classically English turn of phrase, but Ted rejects the premise. Ted says “I disagree, you know? I think it’s lack of hope that comes and gets you. See, I believe in hope.” As all Budding Trends readers know (and there are tens of you around the world), I couldn’t help but think Lasso was channeling the line from Shawshank: “Hope is a good thing, maybe the best of things.”
I suspect those who have known me for a while wouldn’t consider me to be a natural optimist. And years of litigating cases probably hasn’t done anything to change that tendency. I understand why those applicants and would-be patients in the Alabama medical cannabis program would be bereft of hope these days. After all, it has been nearly four years since the Alabama medical cannabis program was enacted by the legislature and not one patient has received medical cannabis.
But somehow in the face of years of advising clients and potential patients going through the hell that can be waiting on lawmakers, regulators, and courts to get a medical cannabis program off the ground, I became an optimist. At first, I’m sure it was little more than putting on a brave face for disappointed, frustrated, and even angry folks wondering why they couldn’t simply get resolution to their dreams. Perhaps this was the “fake it ‘til you make it” phase of my journey, but over time I actually became optimistic that Alabama would be able to launch a medical cannabis program that could provide relief to Alabamians so desperate for a different kind of therapy for what ails them.
I know, maybe as much as anyone in Alabama, how arduous and costly it has been to stand by while this process has played out. I have friends and clients holding licenses issued by the State of Alabama to cultivate and process marijuana. They are wondering whether the countless hours, dollars, and worry have been worth it, and similarly whether it is worth pouring the same into an unknown future. And I have other friends and clients in the integrated and dispensary arena feeling stuck in a regulatory and judicial morass that feels like wading through quicksand.
And, if we’re being honest with each other as friends should, I have left untold fees on the table choosing not to take a side in the long-running litigation because I choose not to take sides when I have clients with different goals and wishes.
For all of these reasons, I am incredibly empathetic to all involved in the seemingly endless delays in the program. But I absolutely believe that Alabama is poised to launch its medical program. Yeah, this is coming from a guy who relies on pop cultural references and jam band quotes. But that guy has never lied to you and has always done his best to take the broader view and make calculated judgments about the facts as they evolve and the future.
I don’t have a crystal ball, and I don’t have a monopoly on predictions. But as I write this, I remain optimistic that Alabama’s medical cannabis program will launch, and it will do so sooner than many believe.
As Coach Lasso instructed: “Onward. Forward.”
Thanks, as always, for stopping by. Your friends at Budding Trends will be right here.
This Week in 340B: February 11 – 17, 2025
Find this week’s updates on 340B litigation to help you stay in the know on how 340B cases are developing across the country. Each week we comb through the dockets of more than 50 340B cases to provide you with a quick summary of relevant updates from the prior week in this industry-shaping body of litigation.
Issues at Stake: Contract Pharmacy; Medicare Payment; Rebate Model
In a case challenging a proposed state law governing contract pharmacy arrangements in Missouri, the court granted in part and denied in part defendant’s and intervenor’s separate motions to dismiss.
In a breach of contract claim filed by a 340B covered entity against several related party Medicare Advantage plans, defendants filed a reply in support of their motion to compel plaintiff’s claims spreadsheets.
In five cases against the Health Resources and Services Administration (HRSA) alleging that HRSA unlawfully refused to approve drug manufacturers’ proposed rebate models:
Five amicus briefs were filed in support of the drug manufacturer.
In four such cases, drug manufacturers filed a joint position statement on consolidation.
In one such case, a drug manufacturer filed a notice of opposition to consolidation and memorandum in opposition to intervenors.
In one such case, the government filed a position statement in support of consolidation.
Kelsey Reinhardt and Nadine Tejadilla also contributed to this article.
Personal Jurisdiction Considerations for International Biosimilars Companies
The Federal Circuit recently issued decisions in a pair of appeals that provide guidance about when international filers of abbreviated Biologics License Applications (aBLAs) are subject to jurisdiction in the United States. Specifically, the Federal Circuit held that international biosimilars companies are subject to jurisdiction in the United States when they have submitted an aBLA with the intent to market the finished product in the forum state.
1. Regeneron’s Patent Infringement Lawsuits
The plaintiff in each case is Regeneron Pharmaceuticals, Inc., which holds Biologics License Application (BLA) No. 125387 for EYELEA®, which is approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with angiogenic eye diseases—Wet Age-Related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), Diabetic Macular Edema (DME) and Diabetic Retinopathy (DR)—via injection into the body of the eye.
Regeneron sued several companies, including Samsung Bioepis Co., Ltd. (SB) and Formycon AG (Formycon), that had filed aBLAs with the FDA seeking approval under the Biologics Price Competition and Innovation Act (BPCIA) to market EYLEA® biosimilars. The cases were consolidated in the U.S. District for the Northern District of West Virginia and the district issued preliminary injunctions against SB and Formycon, barring them from offering for sale or selling the products described in their aBLAs, which have been approved by the FDA. SB appealed the preliminary injunction on several grounds, including that they were not subject to personal jurisdiction, which is the focus of this article.
2. SB’s Connections to the United States
SB is a biosimilar-products company headquartered in Incheon, South Korea. SB argued it has no facilities or employees in the United States; is not registered to do business and has no registered agent in West Virginia; and does not do business with entities in West Virginia. SB also argued that although it would sell its finished product to Biogen MA Inc. (a U.S. company) in a state other than West Virginia, it would not distribute, market or otherwise sell the product in the United States.
3. Formycon’s Connections to the United States
Formycon is a biopharmaceutical company based in Bavaria, Germany. Formycon argued that it has no “direct” ties to West Virginia; is not registered to do business and has no registered agent there; has no assets or employees there; and that it had contracted with manufacturers and packaging partners who would produce the finished product and related materials in other states. Formycon further argued that having developed the product pursuant to an agreement with another German company, it had no plans or rights to itself commercialize the product in the United States. Instead, the product would be sold to another company for marketing and distribution, and Formycon would have no control over the selection of that company or its decisions regarding commercialization.
4. The Federal Circuit’s Jurisdiction Analysis
When evaluating if a defendant is subject to personal jurisdiction in the forum of a particular state, the court looks to (1) whether the state’s long-arm statute permits service of process and (2) whether the assertion of jurisdiction would be inconsistent with due process under the U.S. Constitution. In many states, including West Virginia, the long-arm statutes are “coextensive with the full reach of due process,” so the questions collapse into one constitutional inquiry.
Under the U.S. Constitution, a court in a state may exercise jurisdiction over a defendant that has sufficient “minimum contacts” with the state that it would not “offend traditional notions of fair play and substantial justice.” This standard requires that the defendant’s suit-related conduct create a “substantial connection” with the forum state. The application of the standard in these cases is not necessarily straightforward because patent infringement cases based on an aBLA filing are not easily analogized to other types of actions or even traditional patent infringement cases.
The Federal Circuit, therefore, relied on its precedent in Acorda Therapeutics Inc. v. Mylan Pharmaceuticals Inc., 817 F.3d 755 (Fed. Cir. 2016), which considered the jurisdictional question in the context of a suit arising out of the filing of an Abbreviated New Drug Application (ANDA). In Acorda, the court had held that “minimum contacts” were satisfied by planned future interactions with the state. The submission of an ANDA with the intent to distribute the generic product in a state was held sufficient to support exercising jurisdiction.
Extending Acorda to aBLA cases, the Federal Circuit found similar evidence of conduct sufficient to exercise jurisdiction. Specifically as to SB, the court observed that SB had filed an aBLA; had served Regeneron with a Notice of Commercial Marketing, which communicates an intent to market upon FDA approval; had engaged various partners within the United States; and had entered into a nationwide distribution agreement with a U.S. company, through which SB retained “significant involvement” in commercialization activities.
Notwithstanding the apparent differences in involvement in commercialization activities, the Federal Circuit also found that Formycon intended to market the finished product in West Virginia and other states. As with SB, the court relied on Formycon’s filing of the aBLA, service of Notice of Commercial Marketing, and partnering with U.S. companies to manufacture, package and label its product. Although it had not yet entered into an agreement with a marketing partner, the court found Formycon intended to ultimately distribute the finished product nationwide.
Thus, filing an aBLA, providing Notice of Commercial Marketing, and having more than speculative plans to market the finished product throughout the United States appears sufficient to subject an international biosimilar company to jurisdiction in any state having a long-arm statute coextensive with the U.S. Constitution. The stronger the relationship to commercialization plans, the stronger the argument will be for jurisdiction, although these factors appear to primarily support a finding of jurisdiction as opposed to playing a significant role in the analysis in the first instance.
5. Guidance for International Biosimilars Companies
International biosimilars companies that file aBLAs in the United States with plans to market the finished product should expect a high likelihood of being subject to jurisdiction for related patent infringement cases. Some steps may mitigate the risk, however, and increase the likelihood of avoiding jurisdiction:
Reduce contact with the United States as much as possible. For example, perform all development, sourcing, manufacturing, packaging and labeling outside the United States.
Introduce layers between the aBLA filer and the ultimate marketer. For example, the aBLA filer may contract with other international companies that, in turn, independently contract with a marketing partner in the United States. If the agreement between the aBLA filer and the second international company is not limited to marketing rights in the United States, that may further help.
Carve out particular states. If there are states in which the international biosimilar company does not want to be subject to jurisdiction, expressly exclude those states from commercialization agreements.
These and other factors can significantly affect whether a company is ultimately subject to jurisdiction in the United States, and similar considerations may affect other partners in the international supply chain.
Health Canada Launches FOP Labeling Awareness Initiative
As we have previously blogged about, Health Canada published front-of-pack (FOP) labeling regulations in 2022, which require warnings for most foods high in saturated fat, sugars, and/or sodium. See also Front-of-package nutrition symbol labelling guide for industry – Canada.ca. The regulations will begin to be enforced on January 1, 2026, although the warnings can be voluntarily implemented earlier and have already begun to appear on Canadian shelves.
Recently, Health Canada’s Food and Nutrition Directorate launched an initiative to bring awareness to the new warnings. The initiative aims to inform consumers of the symbol that will be used (black and white box with a magnifying glass, a “high in [X]” declaration, and the words “Health Canada”), its utility (intended to help consumers make informed health choices), and the reason why some pre-packaged foods don’t have it (e.g., the food is a fruit or vegetable or other food exempted because it offers health protection benefits).
We will continue to monitor developments on FOP labeling rules in Canada, the U.S., and other jurisdictions.
President Trump Signs Executive Order Establishing the Make America Healthy Again Commission
On February 13, 2025, President Donald J. Trump signed an Executive Order establishing the President’s Make America Healthy Again Commission. This initiative, chaired by the newly-confirmed U.S. Health and Human Services Secretary Robert F. Kennedy Jr., aims to tackle the root causes of chronic diseases that affect millions of Americans.
According to the order, six in ten Americans have at least one chronic disease, and four in ten have two or more. The commission aims to review the American diet, “absorption of toxic material,” and “food production techniques,” as part of its objectives.
The Commission has outlined four main policy directives to achieve its goals: (1) requiring federally funded research to be transparent; (2) prioritizing researching the root causes of illness; (3) working with farmers to ensure our food supply is healthy and abundant; and (4) increasing the flexibility of health insurance coverage to provide better support for disease prevention.
The composition of the Make America Healthy Again Commission will include the Secretary of Health and Human Services as Chair, and the Assistant to the President for Domestic Policy as Executive Director, and top officials across several federal agencies related to health, the environment, food and drugs, and others.
The EO requires that within 100 days of the order, the commission will provide a preliminary assessment identifying the causes of childhood chronic disease in America.
Alabama Plans to Issue Medical Cannabis Testing License This Summer

What if I told you that you could get a license to test medical cannabis in Alabama this July? Now is that something you might be interested in?
Lost amidst the torrent of confusing messages and general circus surrounding Alabama’s medical cannabis rollout is that the Alabama Medical Cannabis Commission has actually issued licenses to a number of would-be cannabis operators. More than a half dozen cultivators are licensed to grow medical cannabis, and four processors are licensed to process medical cannabis. These companies need testing facilities to analyze their products, and there is currently only one testing facility licensed to do so.
One testing facility is simply not enough to push out the amount of cannabis that will be necessary to operate a statewide medical cannabis program. With a single facility, there will be a huge logjam and a substantial delay in getting medicine to qualifying patients. The AMCC acknowledged as much the day it issued that license and has reiterated that point on multiple occasions since – hence the announcement last week that the AMCC will be offering additional testing licenses in the coming weeks.
Is It Advisable to Pursue a Medical Cannabis Testing License?
First, a word of caution. Because of court injunctions and administrative stays in issuing licenses, no one is currently licensed to dispense medical cannabis. Nor are there any patients licensed to obtain medical cannabis. Without patients and dispensaries, there is not a substantial market for testing medical cannabis. Therefore, anyone interested in pursuing a testing license should do so only after determining they are willing to take the calculated risk that dispensaries will eventually be opened and patients qualified to purchase medical cannabis.
Personally, as I wrote recently, I believe that the court system will soon provide a pathway for exactly that. But I don’t have a crystal ball and anyone considering this path has to determine their own risk tolerance.
I’m a lawyer, so I can tell you about regulations and some of the politics. I can’t tell you it’s going to be a big moneymaker. I do think the first/second mover status could be helpful, but there is no cap on the number of medical cannabis testing licenses under Alabama law, and there could be increased competition for what ultimately will be a limited supply of medical cannabis to be tested.
What Does a Testing License Allow You to Do?
A state testing laboratory license authorizes the licensee to possess and test cannabis and medical cannabis products cultivated or processed at licensed facilities.
There is an annual license fee of $30,000 for medical testing facilities.
The AMCC conveniently provided the following timeline on its website:
2025 State Testing Laboratory License Offering Timeline
Health Agencies Face Terminations; Jim Jones Resigns from FDA’s Human Foods Program
Thousands of workers employed across the Department of Health and Human Services received notices that they would be terminated following four weeks of leave, including at least 89 members of FDA’s Human Foods Program staff, as part of the Trump administration’s overhaul of the federal workforce. The layoffs follow the confirmation of Robert F. Kennedy, Jr. as HHS secretary on February 13.
Terminated staff from FDA’s Human Foods Program include those working on nutrition, infant formula, and food safety response, as well as 10 staff members “who were charged with reviewing potentially unsafe chemicals in the nation’s food supply.”
Jim Jones, FDA’s deputy commissioner for human foods, resigned from the Agency on February 17, citing the “indiscriminate firing” of food program staff and Robert F. Kennedy, Jr.’s rhetoric toward staff. In a letter, Jones wrote: “I was looking forward to working to pursue the Department’s agenda of improving the health of Americans by reducing diet-related chronic disease and risks from chemicals in food. It has been increasingly clear that with the Trump Administration’s disdain for the very people necessary to implement your agenda, however, it would have been fruitless for me to continue in this role.”
Jones, who was appointed as deputy commissioner for human foods in 2023, had committed to priority areas of preventing foodborne illness, decreasing diet-related chronic disease, and safeguarding the food supply, as we previously blogged.