TTB Proposes Rules on Nutrition and Allergen Labeling on Alcoholic Beverages

On January 17, 2025, the Alcohol and Tobacco Tax and Trade Bureau (TTB) proposed two rules that would require nutrition and food allergen labeling on alcoholic beverages, including wines, distilled spirits, and malt beverages. The proposed rules are in response to the Department of the Treasury’s February 2022 report on Competition in the Markets for Beer, Wine, and Spirits, which recommended TTB revive or initiate such rulemaking.
The proposed Alcohol Facts Statements in the Labeling of Wines, Distilled Spirits, and Malt Beverages would require disclosure of per-serving alcohol, calorie, and nutrient content information in an “Alcohol Facts” statement on all TTB-regulated alcoholic beverage labels. Specifically, the Alcohol Facts statement would be required to include:
The serving size of the product;
The number of servings per container;
Alcohol content as a percentage of alcohol by volume;
The mount of pure ethyl alcohol per serving in fluid ounces;
The number of calories per serving; and
The amount of carbohydrates, fat, and protein per serving.
The amount of sugars may be optionally declared in the Alcohol Facts statement. The statement must be presented in a panel or linear format. An example of each format is below:
The proposed Major Food Allergen Labeling for Wines, Distilled Spirits, and Malt Beverages would require labeling disclosures of all major food allergens used in the production of TTB-regulated alcoholic beverages. The declaration must consist of the words “Contains Major Food Allergen(s)” followed by a colon and the name of the food source from which each major food allergen is derived. Processing aids may include the parenthetical explanation “(processing aid)” following the name of the allergen. If major food allergens used in the production of distilled spirits have been completely distilled such that no protein remains, the declaration is not required.
TTB is accepting comments on both proposed rules until April 17, 2025, at regulations.gov. Comments on the nutrition labeling rule should be filed in Docket No. TTB-2025-0002, and comments on the allergen rule should be filed in Docket No. TTB-2025-0003.
FDA Issues New Recommendations on Use of AI for Medical Devices, Drugs, and Biologics
In its most recent effort to keep pace with advancing technology, the US Food and Drug Administration (FDA) recently issued two draft guidances on the use of artificial intelligence (AI) in the context of drugs, biologics, and medical devices.
Medical Device Guidance
The first draft guidance is entitled “Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations.” The guidance provides an overview of the type of documentation and information that companies will need to submit to the FDA to obtain medical device regulatory approval — part of the so-called “marketing submission” process. Among other things, the FDA advises that such documentation and information should include:
A description of the device inputs and outputs, including whether inputs are entered manually or automatically.
A description of how AI is used to achieve the device’s intended purpose.
A description of the device’s intended users, their characteristics, and the level and type of training they are expected to have and/or receive.
A description of the intended use environment (e.g., clinical setting, home setting).
A description of the degree of automation that the device provides in comparison to the workflow for the current standard of care.
A comprehensive risk assessment and risk management plan.
Data management information, including how data were collected, limitations of the dataset, and an explanation of how the data are representative of the intended use population.
A cybersecurity assessment, particularly focusing on those risks that may be unique to AI.
Guidance for Drugs and Biological Products
The second draft guidance issued this month is entitled “Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products.” The guidance addresses considerations for the use of AI to support regulatory decision-making for drugs and biologics. Specifically, the draft guidance discusses the use of AI models to produce information or data to support regulatory decision-making regarding safety, effectiveness, or quality for these products. To that end, the FDA recommends utilizing the following seven-step process to establish and assess the credibility of an AI model output for a specific context of use (COU) based on model risk:
Step 1: Define the question of interest that will be addressed by the AI model.
Step 2: Define the COU for the AI model.
Step 3: Assess the AI model risk.
Step 4: Develop a plan to establish the credibility of AI model output within the COU.
Step 5: Execute the plan.
Step 6: Document the results of the credibility assessment plan and discuss deviations from the plan.
Step 7: Determine the adequacy of the AI model for the COU.
Each of these steps is discussed in detail in the draft guidance, with examples provided. The FDA states that this will provide a “risk-based credibility assessment framework” that will help manufacturers and other interested parties plan, gather, organize, and document information to establish the credibility of AI model outputs when the model is used to produce information or data intended to support regulatory decision-making.
Conclusion
The new guidances from the FDA provide further indication that the agency is closely scrutinizing the use of AI in and in connection with FDA-regulated medical devices, drugs, and biological products. To reduce the risk of unnecessary regulatory delays, companies seeking approval of FDA-regulated products should carefully review their regulatory submissions to ensure they align with the new AI guidance documents.
PFAS Personal Injury Lawsuits: Warning Bells for Users of PFAS
Two recent PFAS personal injury lawsuits have grabbed headlines, as the plaintiffs in the respective lawsuits allege that they developed cancer from public drinking water supplies that were contaminated with PFAS. These are not the only examples of such lawsuits, and while the cases to date target PFAS manufacturers, AFFF manufacturers, the cases are notable because of the allegations that drinking water caused the plaintiffs’ cancers and also, in one lawsuit, that one of the named defendants is a public water utility.
It is not farfetched to imagine a future in the not distant future where PFAS personal injuries expand the scope of defendants to include companies that used PFAS and discharged effluent into waterways that fed public water supplies. It is also not unrealistic to imagine water utilities brought into lawsuits such as the ones described in this article seeking contribution for alleged liability from upstream (literally) companies that discharged PFAS-containing effluent into waterways that the utility had no choice but to accept and send to customers for drinking water.
PFAS Personal Injury Lawsuits and Drinking Water
The two most recent lawsuits alleging cancer as a direct result of ingesting drinking water are Robert Stanfield v. 3M, et al filed in Martin County, Florida and Freeman and Barbara Thompson v. 3M, et al filed in the federal court for Washington state. Both are similar in nature in that they allege that their drinking water sources (private drinking wells in the Thompson case, and a public water supply in the Stanfield case) caused thyroid, prostate and other cancers. Both cases bring claims for compensation against various manufacturers of PFAS and AFFF (firefighting foam) manufacturers, with the Stanfield case adding the Martin County Utilities as a defendant to the lawsuit. In short, the lawsuits allege that the manufacturers of PFAS and AFFF knew of the potential harms to human health associated with PFAS, specifically from contamination to drinking water. The claim against the Martin Cunty Utilities alleges that the water utility knew of the presence and harms of PFAS in drinking water in 2016, yet continued to supply PFAS-contaminated drinking water to customers.
Current and Future Implications To Companies
The current lawsuits present interesting trends in the PFAS personal injury legal space, as they represent a growing trend of personal injury claims alleging drinking water as the cause. The cases are narrowly focused on PFAS manufacturers, AFFF makers, and in some instances, water utilities. This is a trend that will continue in the short term. However, companies must be aware that the future, in my view, the trajectory will look quite different. Plaintiffs will broaden the number of defendants they bring into lawsuits alleging injury from drinking water, to include companies that historically discharged or continue to discharge PFAS-containing effluent into waterways that ultimately feed into drinking water sources. With the ever-increasing reporting obligations for companies under federal and state regulations (including TRI and NPDES permit reporting requirements), the evidence that plaintiffs need to develop cases such as the ones that I predict will be easier to develop.
A head-in-the-sand approach to these issues is unwise for companies. Understandably, companies face significant business interruption issues every day that demand the “here and now” attention of its professionals. Of course, there is always the hope that the day never comes that the company will be brought into a PFAS lawsuit and it is difficult to provide a percent prediction that Board members crave before investing in risk deterrence programs. However, attorneys in the environmental law space and toxic torts space have seen trends like this before. One need not look any further than the asbestos litigation for support of the predicted trend I speak of. More pointedly, in the 1970s and 1980s, the asbestos litigation focused almost entirely on the suppliers of raw asbestos fiber and the companies that manufactured thermal insulation (seen as one of the products with the highest levels of potential exposure, akin to AFFF in the current PFAS litigation). Yet, today, the litigation focuses exclusively on a wide variety of industry and product types that used asbestos as one component of materials such as gaskets, electrical wire, floor tiles, and cosmetics. As the asbestos litigation evolved and a broader and broader net was cast, so too, i believe, will the course of PFAS litigation follow in the footsteps of asbestos litigation. It is for this reason that companies absolutely must prepare now for what is to come.
CMBG3 Law is following judicial, legislative, administrative, and scientific developments relating to PFAS. We represent companies of all sizes on PFAS compliance, litigation, and risk management issues, as well as consult with insurers and financial world firms on PFAS issues. We are recognized thought leaders on the subject of PFAS and are regularly contacted by media – including Bloomberg, Wall Street Journal, Washington Post – for our opinions on PFAS issues.
Federal Circuit Says Proper Orange Book-Listed Patent Must Claim Active Ingredient
In Teva Branded Pharmaceutical Products R&D, Inc. v. Amneal Pharmaceuticals of New York, LLC, the Federal Circuit jumped on the bandwagon of scrutinizing the types of patents that can be listed in the Food & Drug Administration (FDA) Orange Book (Approved Drug Products with Therapeutic Equivalence Evaluations), and decided that the “device” patents at issue were not properly listable. The decision follows recent action in this space by the Federal Trade Commission (FTC), including a general policy statement issued in late 2023, and challenges to more than 100 specific patent listings related to inhaler devices, multidose eyedrop bottles, and autoinjectors.
The Patents at Issue
The patents at issue were five patents Teva had listed in the Orange Book for its ProAir® HFA (albuterol sulfate) Inhalation Aerosol. The patents are described in the Federal Circuit opinion as relating to “improvements in the device parts of inhalers—specifically, the dose counter.” According to the Federal Circuit opinion, “None of the claims … explicitly require the presence of an active drug” as an element of the claimed product.
The ANDA Proceedings and Delisting Counterclaim
Amneal filed an Abbreviated New Drug Application (ANDA) seeking approval for a generic version of Teva’s ProAir® HFA product. Amneal’s ANDA included a paragraph IV certification against the Orange Book-listed patents at issue. After Teva sued for infringement, Amneal filed a counterclaim seeking delisting of the patents at issue. The district court agreed that the patents should not be listed in the Orange Book, but its delisting order was stayed pending Teva’s appeal to the Federal Circuit.
The Federal Circuit Opinion
The Federal Circuit opinion was authored by Judge Prost and joined by Judges Taranto and Hughes.
The Federal Circuit opinion includes a lengthy background section that touches on the origins of the Hatch-Waxman Act, basic principles of the ANDA framework, and statutory and regulatory provisions governing the listing of patents in the Orange Book. For those not familiar with Orange Book listings, it is important to understand that New Drug Application (NDA) holders are required to list certain patents in the Orange Book, but the FDA does not substantively review whether a patent submitted for listing meets the statutory and regulatory listing requirements. In effect, the listing process operates on an honor system, although ANDA filers can challenge Orange Book listings in a counterclaim in paragraph IV litigation as Amneal did here.
Prior to 2021, NDA holders were required to list “any patent which claims the drug for which the applicant submitted the application or which claims a method of using such drug and with respect to which a claim of patent infringement could reasonably be asserted if a person not licensed by the owner engaged in the manufacture, use, or sale of the drug.” In 2021, the Orange Book Transparency Act (OBTA) was enacted to clarify the types of patents that could be listed and codify existing FDA guidance. As amended by the OBTA, an NDA holder must list:
… each patent for which a claim of patent infringement could reasonably be asserted if a person not licensed by the owner of the patent engaged in the manufacture, use, or sale of the drug, and that—
(I) claims the drug for which the applicant submitted the application and is a drug substance (active ingredient) patent or a drug product (formulation or composition) patent; or
(II) claims a method of using such drug for which approval is sought or has been granted in the application.
The Federal Circuit opinion walks through a lengthy analysis to conclude that the patents at issue are not listable because they do not “claim[] the drug for which the applicant submitted the application,” which the court determined was the active ingredient (albuterol sulfate). At the end of its opinion, the court acknowledges but does not “adopt or reject” Amneal’s additional arguments that the patents are not listable because they are not “drug substance” or “drug product” patents under the OBTA.
The Federal Circuit rejected Teva’s arguments that a patent “claims the drug” if the NDA product infringes the claims, explaining in a nine-page analysis that “claiming” and “infringing” have distinct meanings. The court also rejected Teva’s arguments that a patent “claims the drug” if it claims any part of the NDA product, concluding after an eight-page analysis that just because a drug-device combination product was “approved with an NDA” does not mean every part of the product is a “drug.” Rather, according to the Federal Circuit, for drug-device combination products, “the drug for which the application was submitted and approved” is only “the part of the drug-device combination that made it regulatable as a drug in the first place,” i.e., “the active ingredient.”
Using the Definiteness Requirement as a Touchstone For Listability
The definiteness requirement of 35 USC § 112 requires the claims of a patent to “particularly point[] out and distinctly claim[] the subject matter which the inventor … regards as the invention.” Throughout its analysis, the Federal Circuit opinion invokes this definiteness requirement when interpreting the Orange Book listing requirement that the patent “claims the drug for which the applicant submitted the application.” For example, the court emphasizes:
[A] patent claims the drug when it particularly points out and distinctly claims the drug as the invention.
In rejecting Teva’s arguments that the patents would be listable if its claims were construed as requiring the presence of “an active drug,” the court reasoned:
[T]o claim something, a patent must particularly point out and distinctly claim what it purports to be the invention. See 35 U.S.C. § 112(b). And to qualify for listing, a patent must claim at least the active ingredient in the application and the approved drug product. …
In language that may open the door to Orange Book-listing scrutiny of generic claims, the court stated:
A claim requiring the presence of “an active drug” is far too broad to particularly point out and distinctly claim the drug approved in Teva’s NDA. Teva’s construction permits the presence of any active ingredient in any form. As a matter of law, Teva’s construction does not particularly point out and distinctly claim what was approved—the ProAir® HFA with albuterol sulfate as the active ingredient.
Taking Care With Orange Book Listings
In its policy statement, the FTC characterized improper Orange Book listings as potentially anti-competitive behavior, and “put market participants on notice that the FTC intends to scrutinize improper Orange Book listings to determine whether these constitute unfair methods of competition in violation of Section 5 of the Federal Trade Commission Act.” As noted in the Federal Circuit opinion, Amneal also filed antitrust counterclaims, alleging that the improper listings delayed FDA approval of its generic product. Thus, even though Judge Prost opined that “[t]he attractiveness of the thirty-month stay might arguably provide an NDA holder significant incentives to improperly list patents in the Orange Book,” there are good reasons for taking care with Orange Book Listings.
Latest PFAS Consumer Fraud Lawsuit Re-Raises Important Considerations For Companies
On several instances, we have written regarding consumer fraud PFAS class action lawsuits filed in several states. The number of product types targeted for these lawsuits are growing and diverse in terms of the industries targeted. While there has been at least one significant settlement in these lawsuits to date, recently a few of the lawsuits that we previously reported on related to PFAS consumer fraud allegations were dismissed by separate courts.
However, this has not deterred plaintiffs from filing these types of cases, and in fact there are other lawsuits that successfully defeated Motions to Dismiss. The latest PFAS consumer fraud lawsuit targets Samsung this time for PFAS allegedly used in its smart watched. The new lawsuit shows that the number of consumer fraud lawsuits is likely to continue, and consumer goods industries, insurers, and investment companies interested in the consumer goods vertical must pay careful attention to these lawsuits.
Consumer Fraud PFAS Lawsuits – Overview
The consumer fraud PFAS lawsuits filed to date follow a very similar pattern: various plaintiffs bringing suit on behalf of a proposed class allege that companies market consumer goods as safe, healthy, environmentally friendly, etc., or that the companies themselves market their corporate practices as such, yet it is allegedly discovered that certain products marketed with these buzzwords contain PFAS. The lawsuits allege that since certain PFAS may be harmful to human health and PFAS are biopersistent (and therefore environmentally unfriendly), the companies making the good engaged in fraud against consumers to entice them to purchase the products in question.
In the Complaints, plaintiffs typically allege the following counts:
Violation of state consumer protection laws and the federal Magnuson-Moss Warranty Act
Violations of various state consumer protection laws
Breach of warranty
Fraud
Constructive fraud
Unjust enrichment
The plaintiffs seek certification of nationwide class action lawsuits, with a subclass defined as consumers in the state in which the lawsuits are filed. In addition, the lawsuits seeks damages, fees, costs, and a jury trial. Representative industries and cases that have recently been filed include:
Cosmetics industry:
Brown v. Cover Girl, New York (April 1, 2022)
Anderson v. Almay, New York (April 1, 2022)
Rebecca Vega v. L’Oreal, New Jersey (April 8, 2022)
Spindel v. Burt’s Bees, California (March 25, 2022)
Hicks and Vargas v. L’Oreal, New York (March 9, 2022)
Davenport v. L’Oreal, California (February 22, 2022)
Food packaging industry:
Richburg v. Conagra Brands, Illinois (May 6, 2022)
Ruiz v. Conagra Brands, Illinois (May 6, 2022)
Hamman v. Cava Group, California (April 27, 2022)
Azman Hussain v. Burger King, California (April 11, 2022)
Little v. NatureStar, California (April 8, 2022)
Larry Clark v. McDonald’s, Illinois (March 28, 2022)
Food and drink products:
Bedson v. Biosteel, New York (January 27, 2023)
Lorenz v. Coca-Cola, New York (December 28, 2022)
Toribio v. Kraft Heinz, Illinois (November 29, 2022)
Apparel products:
Krakauer v. REI, Washington (October 28, 2022)
Hygiene products:
Esquibel v. Colgate-Palmolive Co., New York (January 27, 2023)
Dalewitz v. Proctor & Gamble, New York (August 26, 2022)
Feminine hygiene products:
Gemma Rivera v. Knix Wear Inc., California (April 4, 2022)
Blenis v. Thinx, Inc., Massachusetts (June 18, 2021)
Destini Canan v. Thinx Inc., California (November 12, 2020)
Latest PFAS Consumer Fraud Case
In Anthony Gonzalez v. Samsung Electronics, the plaintiff alleges that he purchased a Samsung Galaxy Watch to track his fitness goals. The products, plaintiffs argue, were marketed as promoting human health, environmentally sustainable, and suitable for everyday use and wear. Upon testing, the watches were found to have various types of PFAS. Plaintiff alleges that he was therefore deceived by Samsung, and never would have purchased the product if he knew that it contained PFAS. Plaintiff seeks a class certification of all purchasers of the products in question for the time period in question, with a subclass of all purchasers of the products from California.
Recent Rulings In Consumer Fraud PFAS Cases
In California, the Yeraldinne Solis v. CoverGirl Cosmetics et al. case made allegations that cosmetics were marketed as safe and sustainable, yet were found to contain PFAS. The defendants in the lawsuit filed a Motion to Dismiss, arguing in relevant part that the plaintiff had no standing to file the lawsuit because she did not sufficiently allege that she suffered any economic harm from purchasing the product. The plaintiff put forth two theories to counter this argument: (1) the “benefit of the bargain” theory, under which the plaintiff alleged that she bargained for a product that was “safe”, but received the opposite. The court dismissed this argument because the product packaging did not market the product as safe, and the ingredient list explicitly named the type of PFAS found in testing; and (2) an overpayment theory, under which plaintiff alleged that if she knew the product contained PFAS, she would not have paid as much for it as she did. The Court dismissed this argument because the product packaging specifically listed the type of PFAS at issue in the case.
In Illinois, the Richburg v. Conagra Brands, Inc. alleged that popcorn packaging was marketed as containing “only real ingredients” and ingredients from “natural sources”, yet the popcorn contained PFAS (likely from the packaging itself), which was allegedly false and misleading to consumers. The defendant moved to dismiss the lawsuit on several grounds and the Court found in defendant’s favor on one important ground. The Court held that the statements on the popcorn packaging would not mislead an ordinary and reasonable consumer because a consumer would understand “ingredients” to mean those items that are required to be disclosed by the FDA and not materials that may have migrated to the food from the product packaging. In fact, the Court ruled that the FDA “exempts substances migrating to food from equipment or packaging;” and those “do not need to be included in the ingredients list.” The defendant argued that reasonable consumers would not consider PFAS to be an “ingredient” under this regime. In other words, whether or not PFAS migrated into the popcorn, the representations that the popcorn contained “only real ingredients” and “100% ingredients from natural sources” were “correct as a matter of law.” The court dismissed plaintiffs claims on this basis.
Conclusion
Several major companies now find themselves embroiled in litigation focused on PFAS false advertising, consumer protection violations, and deceptive statements made in marketing and ESG reports. The lawsuits may well serve as test cases for plaintiffs’ bar to determine whether similar lawsuits will be successful in any (or all) of the fifty states in this country. Companies must consider the possibility of needing to defend lawsuits involving plaintiffs in numerous states for products that contain PFAS. It should be noted that these lawsuits would only touch on the marketing, advertising, ESG reporting, and consumer protection type of issues. Separate products lawsuits could follow that take direct aim at obtaining damages for personal injury for plaintiffs from consumer products. In addition, environmental pollution lawsuits could seek damage for diminution of property value, cleanup costs, and PFAS filtration systems if drinking water cleanup is required.
While the above rulings are encouraging for companies facing consumer fraud PFAS lawsuits, it is far too early to tell if the trend will continue nationally. As the recent California case shows, plaintiffs continue to file PFAS consumer fraud cases despite the recent dismissals. Different courts apply legal standards differently and these cases are very fact specific, which could lead to differing results. This has been the case in several jurisdictions, where PFAS consumer fraud cases have been permitted to proceed to litigation after initial challenges were made.
It is of the utmost importance that businesses along the whole supply chain in the consumer products industry evaluate their PFAS risk. Public health and environmental groups urge legislators to regulate PFAS at an ever-increasing pace. Similarly, state level EPA enforcement action is increasing at a several-fold rate every year. Now, the first wave of lawsuits take direct aim at the consumer products industry. Companies that did not manufacture PFAS, but merely utilized PFAS in their manufacturing processes, are therefore becoming targets of costly enforcement actions at rates that continue to multiply year over year. Lawsuits are also filed monthly by citizens or municipalities against companies that are increasingly not PFAS chemical manufacturers.
FDA Revokes Authorization for the Use of Red Dye No. 3 in Food and Ingestible Drugs
On 15 January 2025, the US Food and Drug Administration (FDA) announced that it will revoke the color additive authorization for use of FD&C Red No. 3 in food (including dietary supplements) and ingestible drugs. This ban responds to a 2022 color additive petition submitted by several interested parties and filed by FDA in 2023.
In support of the revocation, FDA is relying on the Delaney Clause of the Federal Food, Drug, and Cosmetic Act (21 U.S.C. § 379e(b)(5)(B)), which requires FDA to ban color additives that are found to cause or induce cancer in humans or animals. Specifically, FDA is invoking the Delaney clause as a result of data that shows FD&C Red No. 3 causes cancer in male rats via a sex- and species-specific hormonal mechanism. In fact, according to the preamble of the final rule, “the carcinogenicity of FD&C Red No. 3 was not observed when tested in other animals including female rats and either sex of mice, gerbils, or dogs.” In other words, there is no demonstrable link between consumption of the food additive and cancer in any animal other than male rats, and most importantly, between consumption of the food additive and cancer in humans. The Delaney clause nevertheless requires the revocation of the clearance for FD&C Red No. 3 based on the male rat carcinogenicity data.
Food manufacturers will have until 15 January 2027 to reformulate products containing FD&C Red No. 3, whereas drug producers will have until 18 January 2028. California’s ban on FD&C Red No. 3 in food (along with three other additives) under AB 418 goes into effect a few weeks before FDA’s ban, on 1 January 2027.
McDermott+ Check-Up: January 17, 2025
THIS WEEK’S DOSE
House Committees Organize, Senate Committees Begin Nomination Hearings. House healthcare committees held organizing meetings and announced subcommittee assignments, while Senate committees held nomination hearings for President-elect Trump’s appointees, although none (yet) in the healthcare space.
Senate Committee on Homeland Security & Governmental Affairs Holds OMB Director Nomination Hearing. The Office of Management and Budget (OMB) director confirmation hearing focused on Russell Vought’s previous positions, and mentions of healthcare issues were mostly related to veterans.
Senate Special Committee on Aging Holds Hearing on Improving Wellness Among Seniors. The hearing highlighted programs and policies that can improve seniors’ quality of life.
CMS Announces Next 15 Drugs to be Negotiated in Medicare Part D. The prices for the 15 drugs, which include the anti-obesity medications Ozempic, Rybelsus, and Wegovy, must be negotiated and announced by September 1.
HHS, DEA Issue Two Regulations on Telemedicine Prescribing of Controlled Substances. The regulations, one final and one proposed, from the US Department of Health and Human Services (HHS) and the US Drug Enforcement Administration (DEA) address requirements and pathways for certain providers to prescribe controlled substances via telehealth.
CMS Finalizes NBPP for 2026. The Centers for Medicare & Medicaid Services (CMS) finalized much of what was proposed, including enhanced enforcement against agents and brokers.
CMS Releases Advance Notice for 2026 MA and Part D Payment Policies. The annual Medicare Advantage (MA) and Part D payment update would increase MA revenue by 4.33% in 2026 compared to 2025.
CONGRESS
House Committees Organize, Senate Committees Begin Nomination Hearings. Various committees, including House Ways and Means and House Energy and Commerce, held organizational meetings this week and solidified subcommittee assignments. Reps. Buchanan (R-FL) and Doggett (D-TX) will continue as chair and ranking member, respectively, of the Ways and Means Health Subcommittee. Reps. Carter (R-GA) and DeGette (D-CO) are the new chair and ranking member, respectively, of the Energy and Commerce Health Subcommittee. The Senate Finance and Health, Education, Labor, and Pensions (HELP) Committees have not yet released subcommittee assignments.
The Senate held nomination hearings for Trump appointees this week, including hearings for secretary of defense nominee Pete Hegseth and attorney general nominee Pam Bondi. Nomination hearings for healthcare appointees, including HHS secretary nominee RFK Jr. and CMS administrator nominee Mehmet Oz, are not yet scheduled. RFK Jr. will testify before both the Senate Finance and HELP Committees, although only the Finance Committee will vote to advance his nomination. Committees typically provide a week’s notice before a nomination hearing, so health-related hearings will likely begin no earlier than the week of January 27.
Senate Committee on Homeland Security & Governmental Affairs Holds OMB Director Nomination Hearing. During the hearing, Republicans predominately praised nominee Russell Vought’s previous work as OMB director under Trump’s first Administration and emphasized that they looked forward to working with him again. Democrats pressed Vought on some of his previous positions. With respect to health-policy-focused questions, Democrats asked if he would commit to distributing funds appropriated for SUPPORT Act programs, because they stated that he previously supported withholding funds for funded programs that required reauthorization. Democrats also raised concerns about potential cuts to Veterans Affairs disability benefits.
Senate Special Committee on Aging Holds Hearing on Improving Wellness Among Seniors. The hearing included witnesses from a local police department, research centers, and nonprofits who highlighted that physical and dietary interventions at an earlier age can improve health and longevity and lower costs. Democratic members focused on how lowering prescription drug costs and implementing food programs would benefit seniors, while Republican members focused on addressing financial scams and the costs of implementing programs for older Americans.
ADMINISTRATION
CMS Announces Next 15 Drugs to be Negotiated in Medicare Part D. On the Biden Administration’s last full business day, CMS announced the second round of 15 drugs that will be negotiated in Medicare Part D starting in 2027. Notably, Medicare will negotiate prices for Ozempic, Rybelsus, and Wegovy. Per the Inflation Reduction Act, drugs were selected based on total gross covered prescription drug costs under Medicare Part D. Drug companies with a selected drug will have until February 28 to decide if they will participate in negotiations. However, if a company opts to not participate in the negotiation process, they will face a significant penalty in the form of an excise tax on the sales of that drug, potentially reaching up to 95% of the drug’s U.S. sales.
It is unclear how the incoming Trump Administration will handle both these negotiations and the MA/Part D Technical Rule, released in late November, that proposes to expand coverage of anti-obesity medications in Medicare and Medicaid. Under the Inflation Reduction Act, the final prices for these 15 drugs must be negotiated and announced by September 1, 2025. A fact sheet can be found here, and information about the first round of negotiated drug prices can be found here.
HHS, DEA Issue Two Regulations on Telemedicine Prescribing of Controlled Substances. The agencies released a final rule, Expansion of Buprenorphine Treatment via Telemedicine Encounter, which establishes requirements for the prescription of certain controlled substances via telemedicine and audio-only telemedicine for treatment of opioid use disorder. The final rule requires a DEA-registered practitioner to review the patient’s prescription drug monitoring program data for the state in which the patient is located during an audio-only telemedicine encounter. Additional prescriptions can be issued via other forms of telemedicine as authorized under the Controlled Substances Act, or after an in-person medical evaluation is conducted.
The DEA also released a proposed rule, Special Registrations for Telemedicine and Limited State Telemedicine Registrations, which would establish three special registrations that create a pathway for certain healthcare professionals to prescribe certain controlled substances via telemedicine. The special registration would only apply where the prescribing practitioner has never conducted an in-person medical evaluation of the patient prior to the issuance of the prescription. Comments are due 60 days from publication. For more information on the special registration proposed rule, check out our +Insight.
CMS Finalizes NBPP for 2026. The Notice of Benefit and Payment Parameters (NBPP) finalizes changes to health plans participating on the Affordable Care Act (ACA) Marketplace, as well as new requirements for Marketplaces themselves, agents, brokers, web-brokers, direct enrollment entities, and assisters that help Marketplace consumers. Most proposed policies were finalized and include the following:
Agents and Brokers: CMS enhanced enforcement, including to suspend an agent’s or broker’s ability to transact information with the Exchange. CMS also updated the model consent form that agents, brokers, and web-brokers can use to obtain and document consumer consent.
Grace Periods: CMS will allow health plans to adopt a fixed-dollar payment threshold of $10 or less, adjusted for inflation, under which plans would not be required to trigger a grace period or terminate enrollment for enrollees who fail to pay the full amount of their portion of premium owed.
Failure to File and Reconcile: CMS will require Exchanges to provide notice to consumers and tax filers who have failed to file and reconcile their advanced premium tax credit for two consecutive years.
Plan Options: CMS finalized updates to standardized plan options and non-standardized plan option limits, including requiring issuers to offer multiple standardized plan options within the same product network type, metal level, and service area to better differentiate these plans from one another to reduce the risk of duplicative offerings.
It is unclear how the incoming Trump Administration will handle these policies and whether any will be altered prior to the start of 2026. The final notice was effective January 15, 2025. A fact sheet is available here.
CMS Releases Advance Notice for 2026 MA and Part D Payment Policies. The Advance Notice is released on an annual basis and includes proposed updates to the capitation and risk adjustment methodologies used to calculate payments to MA plans, as well as other payment policies that impact Part D. Key proposals include:
Overall Payment Update: CMS proposed payment updates that would result in an estimated 4.33% increase in MA revenue in 2026 compared to 2025. CMS noted that this percentage translates to an increase of more than $21 billion in MA plan payments from 2025 to 2026.
Risk Adjustment: CMS proposed to complete the three-year phase-in of the Part C Risk Adjustment Model by calculating 100% of the risk scores using only the 2024 CMS-HCC model.
Part C and D Star Ratings: CMS provided a list of eligible disasters for adjustment and lists measures that will be included in the Part C and D improvement measures and Categorical Adjustment Index for the 2026 Star Ratings. CMS is considering additional ways to simplify and refocus the measure set on clinical care, outcomes, and patient experience of care measures, and is considering adding geography to the Health Equity Index reward.
Comments are due by February 10, 2025, which is after President-elect Trump’s inauguration. It is unclear how the incoming Trump Administration will handle the rate notice and whether these policies and payment rates will ultimately be implemented for 2026. The fact sheet can be found here, and a press release can be found here.
QUICK HITS
ASPE Issues Report on Medicare Part D Out-of-Pocket Cap. The HHS Assistant Secretary for Planning and Evaluation (ASPE) found that about 11 million Part D enrollees are expected to reach the $2,000 annual out-of-pocket cap enacted by the Inflation Reduction Act. Read the full report here.
FTC Releases Second Interim Report on PBMs. The Federal Trade Commission (FTC) report on pharmacy benefit managers (PBMs) focuses on specialty generic drug costs and follows the July 2024 first interim report on PBMs. Read the press release here.
HHS Summarizes Public Comments on Consolidation in Healthcare Markets RFI. The report highlights themes from public comments in response to a March 2024 request for information (RFI). The report calls for more ownership transparency and greater disclosures of private equity acquisition activity in healthcare markets; more enforcement action to inhibit mergers and acquisitions; and increased data sharing across federal, state, and local agencies.
CMS Releases Snapshot of Accountable Care Initiatives. The snapshot highlights that 53.4% of traditional Medicare enrollees are in an accountable care relationship in 2025, an increase of 4.3% from 2024. Read the fact sheet here.
CMS Issues Draft 2026 Part D Redesign Program Instructions. The instructions provide information about changes to the structure of the Part D standard benefit that were mandated by the Inflation Reduction Act. Comments are due by February 10, 2025. A fact sheet is available here.
CMS Releases Updated Guidance on Medicaid/CHIP Children’s Continuous Eligibility. The guidance replaces previously issued guidance on the topic, clarifying policies related to implementation in the Children’s Health Insurance Program (CHIP) and for incarcerated youth. The requirement to provide 12 months of continuous eligibility to children under the age of 19 was effective January 2024.
OIG Raises Concerns About FDA Accelerated Approval Pathway. An HHS Office of Inspector General (OIG) report recommended that the US Food and Drug Administration (FDA) modify the accelerated approval pathway to define specific factors that would require the accelerated approval council to advise on certain drug applications, and ensure appropriate documentation of meetings with sponsors in drug approval administrative files.
CMS Releases Guidance on Improving HIV Testing, Prevention, and Care Delivery in Medicaid/CHIP. The guidance provides strategies and opportunities for state Medicaid programs based on the latest scientific evidence and aims to help address access issues raised by two recent OIG reports.
HHS Declares Public Health Emergency, Provides Resources in California. In response to wildfires in southern California, HHS and CMS will provide resources and flexibilities, including extending the Marketplace Open Enrollment period and compiling a Medicaid disaster toolkit for states. Read the press release here.
MedPAC Holds January 2025 Meeting. The Medicare Payment Advisory Commission (MedPAC) meeting included votes on draft recommendations for updating payments for physicians, hospital inpatient and outpatient services, skilled nursing facility services, home health agency services, inpatient rehabilitation facility services, outpatient dialysis services, and hospice services. Sessions also discussed coverage limits on stays in freestanding inpatient psychiatric facilities; cost-sharing for outpatient services at critical access hospitals; and status reports on Part D, MA, and ambulatory surgical center services.
NEXT WEEK’S DIAGNOSIS
President-elect Trump will be inaugurated on January 20. With the new Administration, we expect immediate executive orders and other actions that may impact healthcare. The House and Senate will be in session next week. The Senate HELP Committee will hold an organizational meeting on January 21. Nomination hearings for Trump’s healthcare appointees could begin the week of January 27.
DEA Releases Long-Awaited Telehealth Special Registration Proposal, but Adoption Is Uncertain
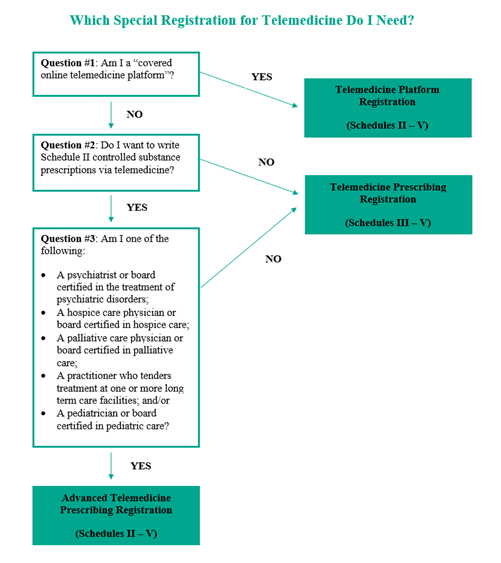
On January 15, 2025, the US Drug Enforcement Administration (DEA) released a proposed rule entitled Special Registrations for Telemedicine and Limited State Telemedicine Registrations. This proposed rule would establish three special registrations, creating pathways for telehealth practitioners to prescribe, and online platforms to dispense, certain controlled substances via telemedicine after flexibilities expire on December 31, 2025. However, it is unclear whether the incoming Trump administration will move forward with the proposed approach for special registration.
IN DEPTH
WHY IT MATTERS
Current federal telehealth-focused controlled substance prescribing flexibilities, initially invoked in response to the COVID-19 public health emergency (PHE), will expire December 31, 2025.
Absent the flexibilities, current law would require telemedicine providers to perform an in-person medical evaluation of a patient prior to prescribing a controlled substance, with certain limited exceptions. One such exception is for providers who hold a “special registration,” the details of which were left within the DEA’s purview. This is the first time the DEA has proposed a special registration since the passage of the Ryan Haight Online Pharmacy Consumer Protection Act of 2008, when it was originally required.
The proposed rule would establish three types of special registrations for telemedicine:
Telemedicine Prescribing Registration, authorizing qualified clinician practitioners to prescribe Schedule III – V controlled substances via telemedicine
Advanced Telemedicine Prescribing Registration, authorizing qualified specialized clinician practitioners (e.g., psychiatrists and hospice care physicians) to prescribe Schedule II – V controlled substances via telemedicine
Telemedicine Platform Registration, authorizing covered online telemedicine platforms, in their capacity as platform practitioners, to dispense Schedule II – V controlled substances.
Special registrants would be required to maintain a State Telemedicine Registration (issued by the DEA) for every state in which the special registrant treats patients, unless otherwise exempted.
The proposed rule would also impose detailed requirements for practice standards, prescription information, and documentation, including requirements related to prescription drug monitoring program (PDMP) checks, use of audio-video technology, restrictions on Schedule II controlled substances, data reporting to the DEA, identity verification, clinician credentialing, and record retention.
The proposed rule was released just days before the incoming Trump administration takes office. Whether the new administration will allow the proposed rule to remain open for public comment or take a different approach remains unclear.
BACKGROUND
Under the Ryan Haight Act, a telemedicine provider is required to perform an in-person medical evaluation of a patient prior to prescribing a controlled substance, with certain limited exceptions. One such exception is for providers who hold a “special registration.” The Ryan Haight Act requires the DEA to establish the circumstances and procedures under which a special registration may be issued. In the more than 16 years since the act’s passage, the DEA has failed to implement such a process, even though Congress imposed a deadline of October 2019 in the 2018 SUPPORT for Patients and Communities Act for the promulgation of final regulations.
In March 2020, in response to the PHE, the DEA invoked flexibilities that allow for prescribing controlled substances via telemedicine without an initial in-person visit. The current extension of the flexibilities, pursuant to a November 2024 rule, authorizes all DEA-registered practitioners to prescribe Schedule II – V controlled medications via telemedicine without an initial in-person examination through December 31, 2025.
Stakeholders had hoped that the DEA would permanently adopt flexibilities for telemedicine prescribing of controlled substances after the PHE, including finally adopting a special registration process. In February 2023, the DEA and the Substance Abuse and Mental Health Services Administration proposed two rules: the general telemedicine rule and the buprenorphine rule. The two proposals would have established additional potential pathways for prescribing certain controlled substances in limited quantities via telemedicine without an initial in-person medical examination while also imposing detailed recordkeeping requirements. Notably, the proposed rules did not include a special registration process for telemedicine providers.
The DEA received a record 38,000 comments in response to the February 2023 proposed rules, including comments from federal lawmakers. Many stakeholders pointed out that the requirement for an in-person evaluation would make it more challenging for certain patients – those facing significant barriers to accessing care without telemedicine – to continue receiving the controlled medications they need. Subsequently, the DEA issued temporary rules in May 2023 and October 2023 extending the telemedicine flexibilities through December 31, 2024, and stated that it anticipated releasing a final rule addressing telemedicine prescription of controlled substances in fall 2024. In November 2024, the DEA further extended the flexibilities through December 31, 2025, stating that the extension would give it time to promulgate proposed and final rules on telemedicine prescribing and “ensure a smooth transition for patients and practitioners that have come to rely on the availability of telemedicine for controlled substance prescriptions.”
THE PROPOSED RULE
The DEA stated that it has determined that the best course of action to ensure patient access to care while maintaining sufficient safeguards to detect and protect against the diversion of controlled substances is to establish and maintain a separate special registration process for telemedicine.
The special registration would only apply where the prescribing practitioner intends to prescribe controlled substances and has not conducted an in-person medical evaluation of the patient prior to the issuance of the prescription. The proposed special registration would not be applicable to practitioner-patient relationships in which there has been a prior in-person medical evaluation of the patient by the practitioner. The special registration also would not apply to the other forms of the practice of telemedicine authorized under the Ryan Haight Act, including those authorized under the 2025 Expansion of Buprenorphine Treatment via Telemedicine Encounter final rule.
THREE REGISTRATION TYPES
The DEA proposes three types of special registrations for telemedicine. To be eligible for a special registration, the applicant would need to demonstrate a legitimate need for a special registration. An applicant for a special registration also would be required to already have one or more DEA registrations to prescribe (if a clinician practitioner) or dispense (if a platform practitioner), unless otherwise exempt.
The Telemedicine Prescribing Registration would authorize qualified clinician practitioners to prescribe Schedule III – V controlled substances via telemedicine.
The DEA determined that clinician practitioners have a legitimate need to prescribe Schedule III – V controlled substances when they anticipate treating patients for whom requiring an in-person medical evaluation prior to prescribing could impose significant burdens on bona fide practitioner-patient relationships (e.g., severe weather conditions, living in remote or distant areas, or having communicable diseases).
The Advanced Telemedicine Prescribing Registration would authorize qualified specialized physicians and board-certified mid-level practitioners to prescribe Schedule II – V controlled substances via telemedicine.
To be eligible for an advanced telemedicine prescribing registration, physicians and board-certified mid-level practitioners would need to demonstrate a legitimate need for a telemedicine prescribing registration, as described above, as well as a legitimate need for the prescribing of Schedule II controlled substances. Balancing concerns for vulnerable populations and the high potential for abuse of Schedule II controlled substances, the DEA determined that only the following seven categories of specialized physicians and board-certified mid-level practitioners have a legitimate need for the advanced telemedicine prescribing registration:
Psychiatrists
Hospice care physicians
Palliative care physicians
Physicians rendering treatment at long-term care facilities
Pediatricians
Neurologists
Mid-level practitioners and physicians from other specialties who are board-certified in the treatment of psychiatric or psychological disorders, hospice care, palliative care, pediatric care, or neurological disorders unrelated to the treatment and management of pain.
Clinician practitioners would be required to furnish information on the special registration application that demonstrates their specialized training – for example, board certification, specialized training, or the percentage of their overall practice that falls within one of the specialized practices). Mid-level practitioners would be required to be board-certified. The DEA seeks input on whether other types of practitioners should be included if they can demonstrate specific training and expertise in managing conditions that are traditionally treated with Schedule II controlled substances, and on alternative methods to ensure that practitioners seeking to prescribe Schedule II controlled substances have the appropriate training and expertise to do so safely.
The Telemedicine Platform Registration would authorize covered online telemedicine platforms to dispense Schedule II – V controlled substances through a clinician practitioner possessing either a telemedicine prescribing registration or an advanced telemedicine prescribing registration.
The DEA notes that the term “dispense” in the Controlled Substances Act means “to deliver a controlled substance to an ultimate user, which includes the prescribing and administering of a controlled substance” and encompasses “not only the physical act of handing out medications, but the broader process of providing medications to patients under the direction of a licensed healthcare provider.” The DEA also notes that by serving as intermediaries for the prescribing of controlled substances, covered online telemedicine platforms qualify as “practitioners” engaged in dispensing.
The DEA proposes to define “covered online telemedicine platform” as an entity that facilitates connections between patients and clinician practitioners via an audio-video telecommunications system for the diagnosis and treatment of patients that may result in the prescription of controlled substances, but is not a hospital, clinic, local in-person medical practice, or insurance provider, and meets one or more of the following criteria:
The entity explicitly promotes or advertises the prescribing of controlled substances through the platform.
The entity has financial interests, whether direct incentives or otherwise, tied to the volume or types of controlled substance prescriptions issued through the platform, including but not limited to ownership interest in pharmacies used to fill patients’ prescriptions or rebates from those pharmacies.
The entity exerts control or influence on clinical decision-making processes or prescribing related to controlled substances, including but not limited to prescribing guidelines or protocols for clinician practitioners employed or contracted by the platform; consideration of clinician practitioner prescribing rates in the entity’s hiring, retention, or compensation decisions; imposing explicit or de facto prescribing quotas; or directing patients to preferred pharmacies.
The entity has control or custody of the prescriptions or medical records of patients who are prescribed controlled substances through the platform.
The DEA states that this definition is intended to limit the special registration requirements to only those direct-to-consumer online telemedicine platforms that play a substantial and integral role as intermediaries in the remote dispensing of controlled substances. The DEA notes that ownership and operation of the online or digital system or platform on which the virtual visit takes place are not mandatory criteria within the proposed definition of a covered online telemedicine platform. Similarly, an entity solely operating a platform or system that merely provides the technological service or conduit for a telemedicine encounter to occur, without the presence of one of the additional four factors, would not constitute a covered online telemedicine platform. The definition of covered online telemedicine platform also explicitly excludes certain types of entities whose primary business operations do not rely on, or center around, telemedicine services, including hospitals, clinics, insurance providers, and local in-person medical practices (defined as medical practices where less than 50% of the prescriptions for controlled substances collectively issued by the practice’s physicians and mid-level practitioners are issued via telemedicine in any given calendar month).
The DEA has determined that covered online telemedicine platforms, in their capacity as platform practitioners, have a legitimate need to dispense Schedule II – V controlled substances when they:
Anticipate providing necessary services to introduce or facilitate connections between patients and clinician practitioners via telemedicine for the diagnosis, treatment, and prescription of controlled substances
Are compliant with federal and state regulations
Provide oversight over clinician practitioners’ prescribing practices
Take measures to prioritize patient safety and prevent diversion, abuse, or misuse of controlled substances.
STATE TELEMEDICINE REGISTRATIONS
The DEA would also require the special registrant to maintain a state telemedicine registration for every state in which the special registrant treats patients, unless otherwise exempted. This registration would be issued by the DEA and not by individual states and would operate as an ancillary credential, contingent on the special registration held by the special registrant.
Both clinician practitioners and online telemedicine platforms would be subject to this requirement.
APPLICATION PROCESS
Creation of Form 224S, Form 224S-M, and Fees
The DEA proposes issuing a new registration application, Form 224S, Application for Special Registration for Telemedicine Under the Controlled Substances Act, tailored for special registrations. The registration would last for three years. The registration fee would be $888 for any one of the three types of special registration. The fee for the platform practitioner state telemedicine registration would be $888 for each state in which a state telemedicine registration is sought; however, the clinician practitioner state telemedicine registration would be discounted to $50 for each state in which the clinician practitioner seeks a state telemedicine registration. The DEA notes in its discussion that fees for the state telemedicine registration for clinician practitioners would be discounted to account for the expected lower volume of telemedicine that would be conducted by clinician practitioners compared to covered online telemedicine platforms.
Registrants would be required to notify the DEA within 14 business days of any modification or changes to the information provided in their original application (Form 224S) via a new form, Form 224S-M. For example, if a clinician holding a special registration began employment with a new direct-to-consumer online telemedicine platform not previously disclosed on the clinician’s original Form 224S, the clinician would be required to submit a Form 224S-M.
Physical Location Requirement
All applicants would be required to designate one of their existing 21 U.S.C. 823(g) registered locations as the registered location/physical address (special registered location) of their special registration. The special registered location would serve as the physical point of contact for DEA inquiries and compliance actions. As discussed below, records arising from telemedicine encounters under the special registration would be required to be maintained at the special registered location.
Additional Disclosures
The applicant would be required to provide certain disclosures and attestations on Form 224S, which the DEA states will “enhance transparency, patient safety, and anti-diversion efforts”:
Platform practitioners applying for the telemedicine platform registration would be required to attest to all employment, contractual relationships, or professional affiliations with any clinician special registrant and online pharmacy, and their respective registration numbers. Likewise, clinician practitioners applying for the telemedicine prescribing registration or the advanced telemedicine prescribing registration would be required to attest to all employment, contractual relationships, and professional affiliations, including but not limited to those with covered online telemedicine platforms (and the respective online telemedicine platform’s telemedicine platform special registration number, if applicable).
Clinician practitioners and platform practitioners would be required to attest that they have devised and are committed to maintaining anti-diversion policies and procedures.
Clinician practitioners applying for the advanced telemedicine prescribing registration would be required to disclose their practice specialties.
For each type of special registration, applicants would be required to attest to their legitimate need on their special registration application.
PRACTICE STANDARDS
Under the proposed rule, registrants would be required to adhere to certain practice standards, such as:
Prescription Origination Within the United States. A clinician special registrant must be physically present in the United States when conducting a telemedicine encounter and issuing a special registration prescription. The clinician also would be required to hold the proper licensure and authorization within the state and territory where the practitioner is located when the telemedicine encounter takes place.
Electronic Prescribing for Controlled Substances (ECPS). All special registration prescriptions must be issued through ECPS.
PDMP Adherence. For the first three years after enactment of the special registration process, clinician special registrants would be required to check the PDMPs for the state or territory where the patient is located, the state or territory where the clinician practitioner is located, and any state or territory with PDMP reciprocity agreements with either the state or territory where the patient is located or the state or territory where the clinician practitioner is located. After three years, all clinician special registrants would be required to verify the identity of the patient and run a nationwide PDMP check of all 50 states and any US district or territory that maintains its own PDMP (referred to as the nationwide PDMP check).
If there is no mechanism to perform the nationwide PDMP check after three years, individual special registrants would continue to be required to perform PDMP checks of the states in the three categories described above. Individual special registrants would only be able to issue special registration prescriptions for Schedule II controlled substances to patients located within the same state as the individual special registrant.
The DEA acknowledges that it is currently unlikely that any one healthcare provider has access to all PDMPs nationwide but recognizes that current efforts to standardize, centralize, and interconnect PDMP data are making headway.
Audio-Video Telecommunications. A clinician special registrant would be required to utilize both audio and video components of an audio-video telecommunications system to prescribe under the special registration framework for every telemedicine encounter, whether for an initial visit or subsequent visit or follow-up.
Schedule III – V Special Registration Prescriptions for Opioid Use Disorder. Clinician special registrants would be allowed to issue special registration prescriptions for, and platform special registrants would be allowed to dispense, Schedule III – V controlled substances approved by the US Food and Drug Administration (FDA) for the treatment of opioid use disorder (OUD) through the use of an audio-only telecommunications system, provided that the treatment was initiated through the use of an audio-video telecommunications system. Currently, the only Schedule III – V narcotic drug approved by the FDA for the treatment of OUD is buprenorphine.
The DEA acknowledges that the Expansion of Buprenorphine Treatment via Telemedicine Encounter final rule allows a DEA-registered practitioner without a special registration to issue a prescription for a Schedule III – V controlled substance approved by the FDA for the treatment of OUD via audio-only or audio-video telemedicine for an initial consecutive six-month supply. Following the initial six-month supply, practitioners may prescribe the controlled substance by other forms of the practice of telemedicine authorized under the Controlled Substances Act (such as pursuant to a special registration) or after conducting an in-person medical evaluation.
Schedule II Controlled Substance Prescriptions. The DEA proposes two requirements for special registration prescriptions for Schedule II controlled substances, indicating that it anticipates imposing one or both requirements based on stakeholder comments.
The first proposed requirement would require that the clinician special registrant be physically located in the same state as the patient when issuing a special registration prescription for a Schedule II controlled substance.
The second proposed requirement would require that the average number of special registration prescriptions for Schedule II controlled substances constitute less than 50% of the total number of Schedule II prescriptions issued by the clinician special registrant in their telemedicine and non-telemedicine practice in a calendar month.
Schedule II Controlled Substance Prescriptions for Minors. In addition to the proposed requirements for Schedule II controlled substances described above, clinician special registrants who are pediatricians or board-certified in pediatric care prescribing Schedule II controlled substances to a minor would be required to prescribe in the presence of the minor’s parent or guardian.
State Law Considerations. When issuing a special registration prescription, a special registrant must comply with the laws and regulations of the state in which the special registrant is located and the state in which the patient is located during the telemedicine encounter.
PRESCRIPTION REQUIREMENTS AND “RED FLAG” CONSIDERATIONS
All prescriptions for controlled substances, whether issued via telemedicine or on the basis of an in-person encounter, are required to include the elements specified in 21 CFR 1306.05(a): signature of the prescriber; issue date; patient’s full name and address; drug details (name, strength, dosage form, and quantity); directions for use; and the practitioner’s name, address, and DEA registration number. The special registration proposed rule would require two additional elements for special registration prescriptions:
The special registration numbers of the clinician practitioner and, if a platform practitioner facilitated the prescription, the platform practitioner
The state telemedicine registration numbers of the clinician practitioner and, if a platform practitioner facilitated the prescription, the platform practitioner (unless exempt from the state telemedicine registration requirements).
The DEA indicates that the inclusion of the special registration number would allow pharmacists to determine if the clinician practitioner has the authority to prescribe a Schedule II controlled substance under the special registration while the inclusion of the state telemedicine registration numbers would allow pharmacists to verify that patients are only being prescribed special registration prescriptions by special registrants authorized to practice in the specific state where the patient is located. The DEA notes that pharmacists occasionally encounter what they may perceive as “red flags” for certain telemedicine prescriptions, which can stem from the nature of telemedicine itself, where patients may receive prescriptions from prescribers located at distances far away (both inside and outside the state where the patient is located). The geographical distance can raise doubts about the legitimacy of the prescription and could lead pharmacists to question its validity and refuse to fill the prescription. The DEA suggests that by verifying state telemedicine registration numbers, pharmacists would receive a level of assurance that a special registration prescription is legitimate when it originates from a prescriber located a significant distance from the patient.
DOCUMENTATION REQUIREMENTS
The special registration proposed rule includes the following documentation requirements:
Patient Verification and Photographic Record. Clinician special registrants would be required to establish and maintain photographic records for patient verification. The DEA would require that these records be maintained for two years from the date of the telemedicine encounter.
If the patient does not consent to their photo being captured, the clinician special registrant (or a delegated employee or contractor under the special registrant’s direct supervision) would be allowed to accept a copy of the patient’s federal or state government-issued photo identification card or other forms of documentation provided by the patient.
Special Registration Telemedicine Encounter Record. Clinician special registrants would be required to maintain a record of the date and time of the telemedicine encounter, the address of the patient during the telemedicine encounter, and the home address of the patient. The DEA would require that these records be maintained for two years from the date of the telemedicine encounter.
Credentialing and Clinician Records. Platform special registrants would be required to maintain and update records related to clinician special registrants with whom they enter and maintain a covered platform relationship, including:
Verification of the clinician special registrant credentials, including but not limited to records on education, training, board or specialty certifications, and special registration number and state telemedicine registration number(s)
The employment contract and any other contract between the platform special registrant and the clinician special registrant
Any disciplinary actions or sanctions, or documentation of complaints, disputes, or incidents involving the practice of telemedicine.
Platform special registrants would be required to maintain and update these records every two years and make them readily available to the DEA.
Data Reporting. Pharmacies dispensing special registration prescriptions would be required to report monthly aggregated special registration prescription data on Schedule II controlled substances and certain Schedule III – V controlled substances. Special registrants would be required to report annually aggregated information about their telemedicine practice, including the number of new patients they treat through telemedicine and the total number of special registration prescriptions for Schedule II controlled substances and certain Schedule III – V controlled substances dispensed for the preceding year.
Recordkeeping at the Special Registration Location. The proposed rule would require that records arising from telemedicine encounters under the special registration framework be kept at the special registered location. The DEA acknowledges that, given telemedicine’s nationwide reach – where a special registrant could serve patients in any state – it would pose an unreasonable administrative burden to require the special registrant to maintain records in every state where telemedicine patients are located.
NEXT STEPS AND INCOMING TRUMP ADMINISTRATION
Stakeholders will have 60 days to comment after publication of the special registration proposed rule in the Federal Register. The DEA encourages input on appropriate implementation timelines, or on-ramps for phased or gradual adoption, to help ensure a smoother transition when the final rule takes effect. Practitioners, pharmacies, and industry stakeholders are encouraged to provide their input on the time necessary to operationalize the proposed requirements.
However, the upcoming administration change may affect when – or if – the special registration proposed rule is adopted. Once in office, President-elect Donald Trump is expected to sign an executive order pausing many of the rules proposed by the Biden administration. It is unclear if this rule will be included. Because this proposed rule is a long-awaited attempt by the DEA to create a special registration, the incoming administration may choose to keep the proposed rule open in order to review public comments on the proposed approach. These comments could help inform future rulemaking. If the proposed rule remains open for public comment, stakeholders should consider providing feedback to help educate and inform the new administration on this approach.
China To Revise Registration Requirements for Foreign Food Facilities
On January 3, 2025, the Chinese General Administration of Customs (GAC) published a draft amendment to the Regulations on Registration and Administration of Overseas Manufacturers of Imported Food (“Registration Regulations,” known as GAC Decree 248)[1] for public comments, due by February 19, 2025. China also notified the World Trade Organization (WTO) of the draft through G/SPS/N/CHN/1324.[2]
In 2021, China issued GAC Decree 248 in 2021, which requires that all overseas food manufacturers exporting food products to China to register with GAC.[3] After three years of implementation, GAC has identified areas for refinement and optimization of the framework, aiming to address practical challenges and enhance the efficiency of the registration process for overseas manufacturers.
Notably, the draft newly introduces a regulatory pathway for overseas facility registration, namely, system recognition, which allows the competent authorities of the manufacturing facility’s home country (region) to obtain recognition from GAC.[4]
Under this new pathway, the competent authorities of recognized countries (regions) may submit a list of recommended manufacturers to GAC. Upon receiving these recommendations, GAC will register the listed enterprises and assign them official registration numbers. It appears that manufacturers may not be required to submit individual applications directly to GAC; however, enterprises from recognized countries may need to coordinate with their competent authorities to ensure their information (e.g., enterprise name, facility address, and contact details) is included in the list submitted to GAC.
This proposed approach represents a significant shift from the current framework under GAC Decree 248, which regulates imported food products based on their risk level and establishes two registration pathways, one for “specified foods” (e.g., milk, meat, aquatic products (typically known as high-risk foods)) and another for “others” (e.g., confectionary and solid beverage). More details concerning GAC Decree 248 can be found in our newsletter: Breaking News: China Imposes New Registration Requirements for All Foreign Food Companies. The draft, however, proposes a classification system at the national level, focusing on whether the competent authorities of the manufacturing facility’s home country (region) can obtain recognition from GAC. Therefore, it is likely that the competent authorities at home countries may have to undertake a higher level of responsibility to supervise the companies exporting foods to China.
On the other hand, the draft also specifies dossier requirements for enterprises whose competent authorities are not recognized by GAC. These enterprises must apply for registration with GAC by themselves or through an agent. We note that the dossier requirements differ based on the category of food involved. Specifically, manufacturers of the specified foods under Catalogue of Foods that Require Official Recommendation Registration Letters (“Catalogue”) will need to provide official inspection reports and recommendation letters issued by their competent authority.
The draft further proposes to update the Catalogue, listing 11 categories of high-risk foods subject to government-recommended registration, along with their corresponding risk assessments. Compared with GAC Decree 248, 8 types of food may no longer be subject to the current recommended registration requirements, covering health foods (including dietary supplements), special dietary foods, unroasted coffee beans and cocoa beans, edible fats and oils, etc. For example, previously, health food manufacturers were required to undergo a government-recommended registration process, [5] which often involved lengthy procedures. Under the proposed revisions, health food manufacturers will be able to process facility registrations by themselves, which will greatly simplify the process, leading to faster market entry and reduced compliance costs. Per GAC’s announcement, the Catalogue will be subject to dynamic adjustments. This flexible approach empowers the authority to adapt quickly to evolving dynamics within the food industry and shifts in the regulatory landscape.
In addition, the draft removes the requirements for overseas manufacturers to reapply for registration in certain scenarios, such as changes to the legal representative or changes to the registration number granted by the country (region). Instead, it specifies that reapplying for registration is necessary when such changes have a significant impact on the enterprise’s sanitation management and control for food safety – for example, in the event of a production site relocation.
Importantly, GAC removes the provision that mandates overseas producers to file their renewal applications three to six months before the registration expires. It also clarifies specific food categories that are exempt from facility registration, including food sent by mail or express delivery, cross-border e-commerce retail items, food carried by travelers, as well as samples, etc. It is worth noting that food additive manufacturers exporting products to China are excluded from the facility registration requirement under Decree No. 248; this remains the same in the draft.
Overall, the proposed changes underscore China’s ongoing efforts to strengthen food safety oversight while simplifying administrative procedures for the registration of overseas manufacturers of imported foods. However, there are certain remaining issues that may require further clarification. For instance, it is unclear how the new system recognition mechanism will integrate with the existing framework under GAC Decree 248. One key question is whether companies that have already registered with GAC still need to coordinate with their competent authorities to be included in the list provided to GAC. Such ambiguity could create challenges for businesses navigating the transition between the current and proposed systems.
[1] http://www.customs.gov.cn/customs/302452/302329/zjz/6297231/index.html; Non-official English translation prepared by United States Department of Agriculture (USDA) is available at: https://apps.fas.usda.gov/newgainapi/api/Report/DownloadReportByFileName?fileName=Amended%20Overseas%20Food%20Producer%20Registration%20Regulation%20Notified_Beijing_China%20-%20People%27s%20Republic%20of_CH2025-0005.pdf
[2] https://eping.wto.org/en/Search/Index?countryIds=C156%2CC764%2CC704%2CC458%2CC360%2CC608%2CC410%2CC392%2CC702%2CC158&viewData=G%2FSPS%2FN%2FCHN%2F1324; All comments should be submitted before March 11, 2025.
[3] https://www.gov.cn/gongbao/content/2021/content_5616161.htm
[4] Such recognition is granted under specific conditions, including (1) accepting and passing the inspection of the food safety management system of the country (region) by GAC; (2) signing a food safety cooperation agreement with GAC; (3) signing a mutual recognition agreement of Authorized Economic Operator (AEO) with GAC; and (4) signing other cooperation agreements or joint statements with Chinese government departments that include food safety cooperation content.
[5] Please note that the term “registration” mentioned in this context pertains solely to overseas facility registration under GAC Decree 248 and does not refer to the “blue hat” registration required for health foods in China.
Hold Your Horses – Cannabis Rescheduling Hearings Stayed, Pending Appeal
In the latest development in a road to rescheduling cannabis from Schedule I to Schedule III under the Controlled Substances Act (“CSA”), on January 13, 2025, in the Matter of Schedules of Controlled Substances: Proposed Rescheduling of Marijuana, DEA Docket No. 1362 Hearing Docket No. 24-44, Chief Administrative Law Judge (“ALJ”) John Mulrooney cancelled the January 21, 2025 hearing on the merits of the Drug Enforcement Agency’s (“DEA”) proposal to reschedule cannabis from Schedule I to Schedule III.
After a request by two private movants (the “Movants”) to remove the DEA from its role as proponent of the proposed reclassification rule was denied, the Movants filed a motion for the ALJ to reconsider its denial of this request. On January 13, 2025, ALJ Mulrooney (i) denied the motion for reconsideration but (ii) granted leave for the Movants to file an interlocutory appeal on the merits of ALJ Mulrooney’s refusal to remove the DEA as proponent of the reclassification. While this Order opens the door on appeal to potentially enable to a private actor to replace the DEA as proponent of the reclassification, the January 13 Order will surely cause further delay in the process of potential rescheduling, evidenced by ALJ Mulrooney’s ordering the Movants and the Government to provide a joint status update 90 days from the issuance of the Order, and every 90 days thereafter.
For those hoping that cannabis would be reclassified before the Trump administration enters office, this is a major disappointment. For those who have been paying attention, this is no surprise, and more of the same.
In a constantly evolving and [still – very] nascent industry like the cannabis industry, one truth has remained: it is a fools errand to try to predict if, when, and how regulatory changes and developments will occur at the federal level. For years, there have been similar questions floated and discussed amongst advisors, operators, and investors in the cannabis industry: “when will cannabis be legalized?”,“when will the SAFE act pass”, “surely Congress will do something, right?”.
Federal action is largely an issue of legislative and regulatory priorities (or, as we have seen, a lack-thereof). Folks can talk and pontificate all they want, but the reality has remained the same: States (at this point, 39 in total, having already passed laws allowing medical marijuana use) are left to fend for themselves, as are the businesses trying to operate with one (if not two) arms tied behind their back.
When President Biden requested in October 2022 for the U.S. Department of Health and Human Services (“HHS”) to “initiate the administrative process to review expeditiously how marijuana is scheduled under federal law”, there was tepid excitement. Hey – the White House is asking HHS to look into this… progress! Then, in August 2023, HHS issued a recommendation to the DEA that cannabis be reclassified from Schedule I to Schedule III under the CSA. At this point, industry participants started to cautiously buy in – maybe – just maybe – this will be the time something actually happens. After all, for business operators, a reclassification to Schedule III under the CSA, would have potentially huge implications – potentially rendering §280E of the tax code inapplicable to cannabis businesses, opening the door for cannabis businesses to deduct various business expenses like any other businesses complying with their state and local laws. And yet, here we are, almost two and a half years later, and the industry is still hoping for change at the federal level.
For operators and investors alike, the reality is simple. Now is not the time to focus on what could happen – or what we hope will happen – at the federal level. Industry participants must continue to focus on what they control: increasing operational efficiency to achieve and maintain profitability.
Environmental Developments to Watch in California in 2025
Contaminants of Concern
Perfluoroalkyl and polyfluoroalkyl substances (PFAS)
In September 2024, California’s legislature enacted two new bills restricting the use of PFAS in consumer products.
AB 347 – This statute gives California’s Department of Toxic Substances Control (DTSC) enforcement authority over existing PFAS restrictions on textile articles (AB 1817), juvenile products (AB 652), and cookware and food packaging (AB 1200) (the “covered products” under the “covered PFAS restrictions”). AB 347 also requires manufacturers of covered products to submit a registration to DTSC by July 1, 2029, pay a registration fee, and submit a statement of compliance to DTSC confirming that each covered product complies with the covered PFAS restriction on the sale or distribution of the product that contains regulated PFAS. DTSC will begin enforcing this legislation after July 1, 2030. Given DTSC is the enforcement authority for the above-mentioned covered products, we expect DTSC to release guidance on interpreting AB 1817, AB 652, and AB 1200 in the future.
AB 2515 – This statute prohibits companies from manufacturing, selling, or distributing menstrual products that contain regulated PFAS. “Regulated PFAS” means PFAS “intentionally added to a product” as of January 1, 2025, and will mean “PFAS in a product at or above a limit determined by the department” beginning January 1, 2027. Like AB 347, AB 2515 requires manufacturers to register with DTSC by July 1, 2029, pay a registration fee, and submit a statement of compliance confirming that menstrual products do not contain regulated PFAS.
We expect DTSC to initiate the rulemaking process for both statutes, which would include regulations regarding accepted testing methods for PFAS levels in menstrual products and third-party laboratory accreditations, and regulations to implement, interpret, and enforce the statutes. Both statutes require DTSC to adopt these regulations before January 1, 2029.
Proposition 65
California’s Safe Drinking Water and Toxic Enforcement Act of 1986, Health & Safety Code Section 25249.5 et seq. (“Proposition 65”) prohibits persons in the course of doing business from knowingly and intentionally exposing individuals to certain listed chemicals above a safe harbor level, where one exists, without first providing a “clear and reasonable” warning to such individuals. (Health & Safety Code § 25249.6). The law applies to consumer product exposures, occupational exposures, and environmental exposures that occur in California. Presently, there are approximately 900 listed chemicals known by the State of California to cause cancer, reproductive harm, or both.
In 2025, we will continue to see developments in the implementation and enforcement of this law, of which manufacturers and retailers selling products in California should be aware.
Vinyl Acetate
On December 19, 2024, the Office of Environmental Health Hazard Assessment’s (OEHHA) Carcinogenic Identification Committee (CIC) voted to list vinyl acetate as a carcinogen under Proposition 65. Vinyl acetate is primarily used in glues, plastics, paints, paper coatings, and textiles. Exposure to the chemical can occur through dermal contact, inhalation, or ingestion.
Vinyl acetate was listed despite industry groups claiming that none of the recognized Proposition 65 authoritative bodies consider the chemical to be a carcinogen. OEHHA published evidence of the carcinogenicity of vinyl acetate, which was used by the CIC to support the listing.
Once listed, businesses have 12 months to provide any required warnings.
Warning Labels
Safe harbor regulations provide examples of long-form and short-form warnings deemed “clear and reasonable,” which, if followed, offer businesses an affirmative defense in the event of enforcement. On December 6, 2024, OEHHA amended Proposition 65 to require companies to add at least one chemical name—or the name of two chemicals, if the warning covers both cancer and reproductive toxicity, unless the same chemical is listed for both endpoints—to the short-form warning on the product label for products manufactured and labeled after January 1, 2028. For example:
“[the warning symbol] WARNING: Cancer risk from exposure to [name of chemical]. See www.P65Warnings.ca.gov.”
OEHHA has authorized the continued used of the earlier short-form warning template (that does not name the chemical) for products manufactured and labeled before January 1, 2028:
“[the warning symbol] WARNING: Cancer – www.P65Warnings.ca.gov.”
Manufacturers and retailers selling products in California containing listed chemicals should review their product labeling protocols, as non-compliance may result in an enforcement action. Some manufacturers have employed generic short-form warnings to forestall enforcement actions without determining whether their products actually exposed consumers to listed chemicals. This practice will not be effective after 2027.
Amended Acrylamide Warning Label
On January 1, 2025, OEHHA’s amendments to acrylamide warning label requirements took effect. The new regulation provides:
Warnings must now contain either:
“WARNING”
“CA WARNING”; or
“CALIFORNIA WARNING.”
The warning must be followed by either:
“Consuming this product can expose you to acrylamide;” or
“Consuming this product can expose you to acrylamide, a chemical formed in some foods during cooking or processing at high temperatures.”
The warning must also be followed by at least one of the following:
“The International Agency for Research on Cancer has found that acrylamide is probably carcinogenic to humans;”
“The United States Environmental Protection Agency has found that acrylamide is likely to be carcinogenic to humans;” or
“The United States National Toxicology Program has found that acrylamide is reasonably anticipated to cause cancer in humans.”
The warning may be followed by one or more of the following:
“Acrylamide has been found to cause cancer in laboratory animals”;
“Many factors affect your cancer risk, including the frequency and amount of chemical consumed”’ or
“For more information including ways to reduce your exposure, see www.P65Warnings.ca.gov/acrylamide.”
The newly amended warning language comes after years of ongoing litigation alleging that the previous warning mandate violated the First Amendment (California Chamber of Commerce v. Rob Bonta (2:19-cv-2019 DJC JDP)). Challengers allege that the warning remains unconstitutional as the state has failed to show that the warnings are purely factual and uncontroversial. As described below, the First Amendment is proving to be an effective defense in some circumstances.
Litigation Update: The Personal Care Products Council vs. Rob Bonta
In recent years, the First Amendment has served as a powerful tool for companies subject to Proposition 65 labeling requirements. A 2025 ruling in The Personal Care Products Council vs. Rob Bonta (2:23-cv-01006) will determine the legality of warning labeling requirements regarding titanium dioxide in consumer products. In 2025, the U.S. District Court for the Eastern District of California is poised to rule on the parties’ motions in the case. If the Court grants the Personal Care Products Council’s (PCPC) summary judgment motion, the ruling will have far-reaching impacts on the enforcement of Proposition 65, bolstering the First Amendment defense to Proposition 65 claims where there is a reasonable scientific debate about the hazards of the listed chemical.
The action was brought in 2023 by PCPC a non-profit association of businesses in the cosmetic and personal care products industry, which sued California Attorney General Rob Bonta in his official capacity.
On June 12, 2024, the District Court issued an Order granting PCPC’s request for a preliminary injunction enjoining Bonta and all private enforcers of Proposition 65 from filing new lawsuits to enforce the law’s warning requirement for exposures to titanium dioxide. The District Court agreed with PCPC that the “Prop 65 warning requirements for Listed Titanium Dioxide are not purely factual because they tend to mislead the average consumer” since the warnings may convey a “false and/or misleading message that Listed Titanium Dioxide causes cancer in humans or will increase a consumer’s risk of cancer.” This, according to the District Court, renders PCPC likely to prevail on the merits of its First Amendment claim under Zauderer v. Off. of Disciplinary Couns. of Supreme Ct. of Ohio, 471 U.S. 626 (1985) (government may compel commercial speech so long as it is reasonably related to substantial governmental interest, purely factual, noncontroversial, and not unjustified or unduly burdensome).
PCPC’s pending summary judgment motion was filed on September 10, 2024. If granted, this will be the third case successfully challenging Proposition 65 warnings on First Amendment grounds, with previous cases involving designated glyphosate and acrylamide. See Nat’l. Assoc. of Wheat Growers v. Bonta, 85 F.4th 1263 (9th Cir. 2023); Cal. Chamber of Comm. v. Bonta, 529 F. Supp. 3d 1099 (E.D. Cal. 2021).
Here, the District Court’s June 12, 2024 ruling dramatically halted the prosecution of countless pending claims against cosmetic companies and retailers of cosmetics. A favorable ruling for PCPC in 2025 may embolden companies subject to Proposition 65 requirements to bring an array of constitutional challenges with respect to other designated chemicals, specifically businesses selling products containing a designated chemical where the underlying scientific basis for its designation is controversial. The District Court’s language strongly casts doubt on the constitutionality of “misleading” Proposition 65 labels that lack an adequate scientific basis.
Extended Producer Responsibility (EPR) and Recycling
California continues to pave the way for EPR laws that affect various products. Rulemaking efforts will continue through 2025.
AB 863 – Carpets
Governor Newsom approved AB 863 on September 27, 2024, governing carpet recycling in California. California enacted its first carpet stewardship law in 2010 and has since amended it multiple times. The latest law maintains several basic facets and updates the governance structure of California’s current carpet stewardship program but nominally converts it to a carpet producer responsibility program following the expiration of the current 2023-2027 five-year carpet stewardship plan. The new law punts many specifics of the new program to the discretion of CalRecycle, including performance standards and metrics, key definitions, deadlines, and grounds for approving or revoking an approved plan. CalRecycle must adopt implementing regulations effective no earlier than December 31, 2026. The law purports to deem CalRecycle’s adopted “performance standards” as immune from judicial review under the California Administrative Procedure Act. The law also calls for certain amendments to the existing carpet stewardship plan to be proposed and adopted sooner.
The new law requires all carpet producers doing business in California to form and register with a single producer responsibility organization (PRO). The law requires the PRO to develop a producer responsibility plan for the collection, transportation, recycling, and safe and proper management of covered products in California, along with related public outreach regarding the plan; review the plan at least every five years after approval; and submit annual reports to CalRecycle. An approved plan must be in place within 24 months of the effective date of CalRecycle’s regulations under the new law, which may result in a deadline as early as December 31, 2028. All reports and records must be provided to CalRecycle under penalty of perjury. The law restricts public access to certain information collected for the purpose of administering this program.
The PRO must establish and provide a covered product assessment to be added to the purchase price of a covered product sold in the state by a producer to a California retailer or wholesaler or otherwise sold for use in the state. Each retailer and wholesaler is then required to add the assessment to the purchase price of all covered product sold in the state. This assessment of carpet sales in California parallels existing law. The new law does not specify any other available funding methods for implementing its requirements. The new law also requires the PRO to pay fees to CalRecycle, not to exceed CalRecycle’s actual and reasonable regulatory costs to implement and enforce the program. It further newly requires all carpet sold in California to contain 5% of post-consumer recycled carpet content by 2028, and grants CalRecycle authority to set new rates for 2029 and beyond.
Additionally, the new law requires carpet producers to provide additional information to CalRecycle regarding California carpet sales and compliance with the requirements of an approved plan. CalRecycle must post on its website a list of producers that are in compliance with the requirements of the program. The existing carpet stewardship plan must be amended to allocate 8% of collected assessments to unions for apprenticeship program grants. Compared to current law, penalties for violations increase from $5,000 per day to $10,000 per day, and from $10,000 per day to $25,000 per day if the violation is intentional, knowing, or negligent. CalRecycle may audit a carpet stewardship organization and individual producers annually The law also clarifies that a carpet stewardship organization cannot delegate decision-making responsibility regarding a carpet stewardship plan to a person who is not a member of the organization’s board.
SB 707 – Textiles
In September 2024, California’s legislature enacted the first, and only current, statewide EPR textile program in the U.S. with the Responsible Textile Recovery Act of 2024. The Act requires qualified producers of apparel or textile articles to form and join a PRO that CalRecycle will approve by March 1, 2026. All eligible producers must join the PRO by July 1, 2026. Once formed, the PRO must submit a statewide plan for the collection, transportation, repair, sorting, recycling, and the safe and proper management of covered clothing and textiles to CalRecycle for review. Once the plan is approved, retailers, importers, distributors, and online marketplaces will not be permitted to sell, distribute, offer for sale, or import a covered product into the state unless the producer of the covered product is listed as in compliance. The PRO will charge each participant-producer annual fees for its operation.
By July 1, 2030, or upon approval of the plan, whichever occurs first, noncompliant producers of covered products will be subject to administrative civil penalties up to $50,000 per day.
The Act directs CalRecycle to adopt regulations to implement its provisions with an effective date of no earlier than July 1, 2028. The rulemaking process will be carried out in accordance with California’s Administrative Procedure Act, which provides opportunities for the public, including industry representatives, to shape the policy going forward. Rulemaking efforts associated with SB 707 are not yet listed on CalRecycle’s website, but given the short deadlines imposed by the Act, we can expect updates in the near future.
AB 187 – Mattresses
California’s legislature established the Used Mattress Recovery and Recycling Act (Mattress EPR Act) in 2013 and most recently updated it in 2019. The Mattress EPR Act, which CalRecycle administers, applies to manufacturers, renovators, distributors, and retailers that sell, offer for sale, or import a mattress into California. At least once every five years, the mattress recycling organization reviews the plan for the recovery and recycling of used mattresses and determines whether amendments are necessary. Each year, CalRecycle, through the Mattress Recycling Council, posts lists of compliant manufacturers, renovators, and distributors on its website. If the manufacturer, brand, renovator, or distributor is not on this list, no retailer or distributor may sell a mattress in the state until the department affirms they are in compliance.
CalRecycle may impose an administrative civil penalty of not more than $500 per day on any manufacturer, mattress recycling organization, distributor, recycler, renovator, or retailer violating the Mattress EPR Act. However, if the violation is intentional, knowing, or reckless, the department may impose an administrative civil penalty of not more than $5,000 per day.
SB 551 – Beverage Containers
SB 551, or the California Beverage Container Recycling and Litter Reduction Act, took effect on September 29, 2024 as an urgency statute, necessary for the immediate preservation of the public peace, health, or safety within the meaning of Article IV of the California Constitution. Plastic beverage containers sold by a beverage manufacturer must contain a specified average percentage of post-consumer recycled plastic per year. Manufacturers of beverages sold in a plastic beverage container subject to the California Redemption Value fee must report to CalRecycle certain information about the amounts of virgin plastic and post-consumer recycled plastic used for those containers for sale in California in the previous calendar year. The law authorizes certain beverage manufacturers to submit a consolidated report to CalRecycle with other beverage manufacturers, in lieu of individual reports, if those beverage manufacturers share rights to the same brands or the products of which are distributed, marketed, or manufactured by a single reporting beverage manufacturer. This consolidated report must be submitted under penalty of perjury and pursuant to standardized forms prescribed by CalRecycle.
SB 54 – Plastics and Packaging
At the start of this year, CalRecycle was required to adopt any necessary regulations to implement and enforce its Plastic Pollution Prevention and Packaging Producer Responsibility Act (SB 54). SB 54 imposes EPR on “producers” of packaging materials for achieving the source reduction, recyclability or composability, and recycling rates for their products. Producers may comply with SB 54’s requirements by either joining the Circular Action Alliance (CAA), the PRO selected by the state to administer SB 54, or through assuming individual responsibility for compliance.
CalRecycle met its regulation deadline under SB 54 by publishing the Source Reduction Baseline Report on December 31, 2024, followed by updates to the list of Covered Material Categories regulated by SB 54 on January 1, 2025. The updates to the Covered Material Categories include an increase in materials considered to be “recyclable” or “compostable” while the Source Reduction Baseline Report establishes a baseline measurement for the Department and CAA to define source reduction targets, develop plans and budgets, and the track progress of SB 54’s implementation.
On January 1, 2025, SB 54’s prohibition on the sale, distribution, or importation of expanded polystyrene (EPS) food service items—unless the producer can demonstrate that all EPS used in the state meets a recycling rate of least 25%—went into effect. EPS food service producers may now be subject to notices of violation from CalRecycle and enforcement of penalties for noncompliance of up to $50,000 per violation, per day. Recycling rate mandates for plastic-covered materials do not go into effect until 2028.
SB 1143 – Paint
In September 2024, California enacted SB 1143, which expands the state’s existing Architectural Paint Recovery Program to include a wider range of paint products. “Paint product” is now defined to include interior and exterior architectural coatings, aerosol coating products, nonindustrial coatings, and coating-related products sold in containers of five gallons or less for commercial or homeowner use.
The law tasks CalRecycle with administering the program and approving a stewardship plan for the newly covered paint products. Retailers, importers, distributors, and online marketplaces will be prohibited from selling, offering for sale, or importing these products in California unless the producers are in compliance with the stewardship plan. Producers may comply with SB 1143 requirements by either joining PaintCare, the only recognized paint stewardship organization representing paint manufacturers in California, or through assuming individual responsibility for compliance.
All eligible products must comply with the new requirements by January 1, 2028, or an earlier date set by an approved stewardship plan. By July 1, 2030, or upon approval of the plan, whichever comes first, noncompliant producers will face administrative civil penalties up to $50,000 per day.
Climate Regulation
SB 261 and SB 253
After a year of uncertainty driven by budget constraints, California seems poised to implement its climate disclosure laws (SB 261 and SB 253) that were first passed in 2023. In September 2024, the Legislature passed SB 219, which granted the California Air Resources Board (CARB) a 6-month extension to issue the requisite rules that must be adopted by no later than July 1, 2025. CARB is responsible for administering SB 261 and SB 253.
On December 16, 2024, CARB posted an Information Solicitation that calls for public comments on the implementation of the laws and related issues. The Information Solicitation also invites input on key aspects of the climate disclosure framework that have been subject to speculation since the laws were enacted, such as the definition of “entity that does business in California” (clarifying the cohort within the scope of the laws); the methods for measuring and reporting scope 1, scope 2, and scope 3 emissions; and third-party verification and assurance requirements. The deadline to submit comments through CARB’s website is February 14, 2025.
Cal Chamber v. CARB
On January 30, 2024, the U.S. Chamber of Commerce and other business groups filed Chamber of Commerce of the United States of America et al. v. California Air Resources Board (CARB) et al., No. 2:24-cv-00801 (C.D. Cal. 2024) challenging SB 253 and SB 261 for violation of the First Amendment, the Supremacy Clause, and the U.S. Constitution’s limitations on extraterritorial regulation, including the dormant Commerce Clause.
Regarding the First Amendment facial challenge, the Plaintiffs alleged the laws “compel companies to publicly express a speculative, noncommercial, controversial, and politically-charged message that they otherwise would not express.” Concerning the Supremacy Clause, they argued that by requiring companies to make speculative public statements about emissions and climate-related financial risk, the laws enable “activists and policymakers to single out companies,” pressuring them to reduce emissions within and outside California. As for the constitutional claims, the Plaintiffs alleged that California lacks authority to regulate greenhouse gas emissions outside of the state and that the laws are invalid under the U.S. Constitution’s limitations on extraterritorial regulation because they heavily intrude on Congress’s authority to regulate interstate and foreign commerce.
To expedite the District Court’s ruling, the Plaintiffs moved for summary judgment on the First Amendment challenge. Simultaneously, CARB moved to dismiss the Plaintiffs’ Supremacy Clause and extraterritorial regulation claims. On November 5, 2024, the District Court denied the Plaintiffs’ motion. The Court held that the First Amendment applied to SB 253 and 261; however, it concluded that the constitutional challenge involves factual questions that go beyond pure legal analysis and thus, completing a “fact-driven task” was necessary to decide which of the laws’ applications violate the First Amendment. It held that further discovery is required to complete this “fact-driven” task.
The District Court indicated that it would address CARB’s motion to dismiss in a separate order. That motion is pending as of the date of this publication.
SB 1383 – Organic Waste & Food Collection
Since CalRecycle adopted regulations implementing SB 1383, California communities have made progress in diverting and reducing the disposal of organic waste and thereby reducing the amount of methane emissions from landfills. According to California’s Short-Lived Climate Pollutant Reduction Strategy, 93% of jurisdictions with requirements for collection reported having residential organics collection, and 100% of California communities expanded programs to send still-fresh, unsold food to Californians in need, reducing the waste large food businesses send to landfills every year. Through SB 619, 126 jurisdictions have been granted additional time to comply with SB 1383 regulations.
While progress has been made, local jurisdictions continue to struggle to meet the law’s mandates (namely, reduce organic waste disposal by 75% and reduce edible food waste by 20% by 2025). Rather than revising those mandates or pausing the implementation of SB 1383 to ensure jurisdictions weren’t sanctioned for missing implementation deadlines, the legislature enacted a number of laws to address some of the concerns raised by the regulated community. These include SB 2902, AB 2346, and SB 1046.
SB 2902 extends the rural jurisdiction exemption to comply with organics collection and procurement requirements until January 1, 2027. AB 2346 allows jurisdictions to count specified compost products toward their goals and adopt a five-year procurement target instead of annual goals, and SB 1046 directs CalRecycle to create a programmatic environmental impact report for small to medium composting facilities, aiding local governments and composters by streamlining permitting.
Although CalRecycle initiated formal enforcement actions in 2024, there is no indication that the agency has fined or sanctioned any jurisdiction for non-compliance. As the 2025 target date has now passed, expect enforcement efforts to increase in the months and years ahead.
Energy Efficiency Standards
As covered in our December 10, 2024 news alert, manufacturers and sellers of consumer products in California should be aware that the California Energy Commission continues to bring more enforcement actions and assess large civil penalties for violations of its Title 20 Appliance Efficiency Program. At a time when federal appliance efficiency standard enforcement is expected to recede due to the recent presidential election and imminent transition, California enforcement is likely to continue to grow. Regulated businesses, therefore, should pay increasing attention to Title 20 compliance, not only to avoid large fines but also to ensure continued access to their products in the lucrative California market.
Stationary Source Regulation
AB 1465 – Air Quality Management Districts (AQMDs) Granted Authority to Seek Triple Penalties
For years, the penalty ceilings in California’s Health & Safety Code have limited the ability of California’s regional AQMDs to collect civil penalties for rule violations. Starting January 1, 2025, AB 1465 tripled these ceilings. For example, the typical maximum penalty for strict liability violations—previously $12,090 per violation—has escalated to $36,270 per violation. The new law also requires that air districts (or a court) consider items like health impacts and community disruptions when evaluating penalty amounts (in addition to other factors required to be considered by law). These elevated ceilings only apply to stationary sources that have a Federal Clean Air Act Title V permit and emit certain defined compounds. How air districts will wield this new authority has yet to be seen, but we expect to see increasing penalties for many sources as a result.
Indirect Source Rules Will Continue to be a Hot Topic
While regional air districts are generally limited in their legal authority to regulate mobile sources (that authority is reserved for California’s state air regulator, CARB), indirect source rules (regulation of stationary sources that attract emissions from mobile sources) have received renewed attention as a means by which air districts seek to curb air pollution. With the incoming Trump administration signaling its intent to limit California’s ability to regulate mobile sources, air districts will likely be incentivized to find creative ways to indirectly regulate mobile sources within their districts.
In 2024, the South Coast AQMD received U.S. Environmental Protection Agency (EPA) approval to include such an indirect source rule (ISR) for warehouses as part of its state implementation plan. South Coast AQMD also adopted an ISR in 2024 applicable to rail yards and has been working on a rule applicable to ports for years, which it promises to bring before its board for approval in 2025.
Perhaps observing the South Coast AQMD’s recent ISR adoptions, the Bay Area AQMD also included an ISR in its 2025 rulemaking forecast. However, exactly what such a rule for this district might look like or what source it might seek to regulate remains to be seen.
New National Ambient Air Quality Standard for PM 2.5 Will Likely Drive Rulemaking Activity
California’s major regional air quality districts (the Bay Area AQMD, the South Coast AQMD, and the San Joaquin Valley Air Pollution Control District) have jurisdiction over areas considered to be in non-attainment of national standards regarding particulate matter (PM) 2.5. Areas in persistent non-attainment status risk federal sanctions and the loss of federal highway funding. In early 2024, EPA tightened the PM 2.5 standards even further. As a result, some air districts may consider rulemakings designed to reduce PM2.5 pollution within their jurisdictions. Given that mobile sources are a major contributor of this pollutant, ISR options may become even more appealing in 2025 and beyond.
Mobile Source Regulation
The Clean Air Act preempts states from adopting their own emission standards for new motor vehicles and new motor vehicle engines. However, Section 209 of the Clean Air Act allows California to set its own emissions standards if EPA grants a waiver from the federal preemption or EPA authorizes California to enforce its own standards despite the preemption. In the past year, CARB submitted requests for waiver or authorization for several regulations.
Advanced Clean Fleets Regulation – This regulation applies to trucks performing drayage operations at seaports and railyards; fleets owned by State, local, and federal government agencies; and high-priority fleets that are entities that own, operate, or direct at least one vehicle in California and that have either $50 million or more in gross annual revenue, or that own, operate, or have common ownership or control of a total of 50 or more vehicles. The regulation imposes restrictions on purchasing internal combustion engines, requires fleet owners to phase in zero-emission vehicles (ZEVs) or near-ZEVs beginning in 2024, and imposes reporting and recordkeeping requirements on fleet owners and operators. On January 13, 2025, CARB withdrew the request for waiver and authorization. In a response letter, EPA stated that it, therefore, “considers the matter closed.”
In-Use Locomotive Standards – The regulation has four primary, interrelated components: (1) imposes restrictions on the operation of any locomotive that is “23 years or older” from the original engine build date unless the locomotive exclusively operates in zero-emission configuration within California; (2) requires railroads to make annual deposits into a “Spending Account” based on the locomotive’s emissions in California in the prior year and imposes restrictions on the use of funds in the “Spending Account”; (3) imposes idling requirements that would regulate a locomotive’s function and maintenance; and (4) imposes registration, reporting, and recordkeeping requirements, including the requirement to annually report emissions information for non-zero emissions locomotives. On January 13, 2025, CARB withdrew the request for waiver and authorization. By response letter, EPA stated it therefore “considers this matter closed.”
Amendments to the Small Off-Road Engines Regulations – The amendments include improvements to evaporative emissions certification procedures, revise the compliance testing procedure, update the evaporative emissions certification test fuel to represent commercially available gasoline, and align aspects of the regulation requirements with the corresponding federal requirements. EPA granted the authorization request on December 19, 2024.
The “Omnibus” Low NOx Regulation – The regulation establishes the next generation of exhaust emission standards for nitrogen oxides (NOx), PM, and other emission-related requirements for new 2024 and subsequent model year on-road medium- and heavy-duty engines and vehicles. EPA granted the authorization request on December 17, 2024.
Advanced Clean Cars II Program – The regulations amend the Zero-emission Vehicle Regulation to require an increasing number of ZEVs and amends the Low-emission Vehicle Regulations to include increasingly stringent particulate matter, Nox, and hydrocarbon standards for gasoline cars and heavier passenger trucks to continue to reduce smog-forming emissions. EPA granted the authorization request on January 6, 2025.
Amendments to California’s In-Use Off-Road Diesel-Fueled Fleets regulation – The amendments will require fleets to phase out use of the oldest and highest polluting offroad diesel vehicles in California, prohibit the addition of high-emitting vehicles to a fleet, and require the use of R99 or R100 renewable diesel in off-road diesel vehicles. EPA granted the authorization request on January 3, 2025.
If the current EPA administration does not grant the pending waiver requests, then it is unclear how EPA under the Trump administration will decide on the waiver requests. Our November 6, 2024 news alert discusses these waiver issues in more detail.
CARB also enacted the zero-emission forklift regulation on August 2, 2024. The regulation accelerates the transition towards zero-emission forklifts by restricting fleet operators/owners from owning, possessing, and operating Large Spark Ignition (LSI) forklifts starting on January 1, 2026, and requiring fleet operators to phase out Class IV LSI forklifts of any rated capacity, as well as Class V LSI Forklifts with rated capacity less than 12,000 pounds according to the compliance schedule in the Regulation. These forklifts will need to be phased out by January 1, 2038.
Cal/OSHA Developments
Cal/OSHA Lead Exposure Regulations
The California Division of Occupational Safety and Health’s (Cal/OSHA) updated lead standards, which were approved on February 15, 2024, and went into effect on January 1, 2025. These apply to both general and construction worksites and replace standards that are decades old, based on data from over 40 years ago. The amended standards modify the permissible exposure limit (PEL), action level (AL), workplace hygiene practices, and medical surveillance requirements relating to lead in the workplace.
The reduction of the PEL and AL is significant; the threshold that triggers various regulatory requirements is now considerably lower. Many new industries will likely be covered. The PEL is now 10 µg/m3 (8-hour-time weighted average), an 80% reduction from the earlier PEL (50 µg/m3). The AL is now 2 µg/m3, a 93% drop from the prior AL (30 µg/m3).
Regulations for General Industry now define certain tasks as “Presumed Significant Lead Work” (PSLW). Until employers perform an employee exposure assessment, they are required to provide employees performing PSLW with interim protections.
For the construction industry, the regulations also define various “trigger tasks” levels, which assume a certain level of employee exposure. These “triggers” require protective measures for employees performing these tasks until an employee exposure assessment is completed.
Cal/OSHA Silica Emergency Temporary Standard
Cal/OSHA stated that California is experiencing a “silicosis epidemic” among artificial stone fabrication workers. In December 2023, the Occupational Safety and Health Standards Board (OSHB) approved the Emergency Temporary Standard (ETS) on Respirable Crystalline Silica (RCS) in response to these circumstances. The ETS intends to protect employees working with artificial and natural stone containing more than 10% crystalline silica. Additional protections apply to workers performing “high exposure trigger tasks.”
On December 19, 2024, OSHB voted unanimously to make the Silica ETS permanent. The decision is a step towards making these emergency measures permanent. The current proposal continues the protections the ETS has introduced, with some changes. These include a new medical removal subsection and updates to the medical surveillance subsection.
The proposed medical removal provisions provide protections to employees when a physician or other licensed healthcare professional (PLHCP) recommends that they be removed from a job assignment or that the job be modified to reduce exposure to RCS. The proposed updates to the medical surveillance provisions include specific medical procedures to be conducted for the required initial and periodic examinations. PLCHPs and specialists would also be required to submit certain information to the California Department of Public Health for each silica medical examination conducted.
The Office of Administrative Law has 30 days to approve or deny the proposal. We expect a decision in mid-January 2025.
Cal/OSHA Increases Staffing for Its Bureau of Investigations Unit
In August 2024, Cal/OSHA announced that it had increased staffing for its Bureau of Investigations (BOI) unit. Cal/OSHA says this would “allow BOI to tackle more cases and ensure that the most negligent of employers are held accountable.”
The BOI is responsible for investigating employee fatality and serious injury cases, and preparing and referring cases to local and state prosecutors for criminal prosecution. Cal/OSHA was criticized in early 2024 for the short-staffed status of BOI. Given the recently enhanced staffing, employers should expect that BOI investigations will likely increase in 2025.
Bird Flu
On December 18, 2024, Governor Newsom declared a state of emergency for Avian influenza (H5N1) (“bird flu”) in California. On December 27, 2024, the Division of Workers’ Compensation (DWC) advised employers and healthcare professionals to look for occupational cases of bird flu. There have been no cases of human-to-human transmission in California—nearly all affected persons had exposure to infected cattle. In light of DWC’s recommendations, employers should nevertheless review Cal/OSHA’s guidance on bird flu for employers.
Water Rights, Tribal Issues, Public Lands, Endangered Species
Threatened Species Listing of Monarch Butterfly
On December 12, 2024, the U.S. Fish and Wildlife Service (FWS) proposed listing the monarch butterfly as a threatened species with a special section 4(d) rule under the Endangered Species Act (ESA). The special 4(d) rule would provide very narrow exemptions to the ESA’s broad prohibition on unauthorized take for certain types of activities that may otherwise impact the species. FWS also proposed designating nearly 4,500 acres in California as critical habitat that would extend from the California Bay Area’s Marin County down the state’s western coast to Ventura County north of Los Angeles.
If finalized as proposed, this listing would stand as the largest listing decision in ESA history, affecting the entire lower forty-eight states. FWS is receiving public comment through March 12, 2025.
Central Valley Project and State Water Project
The U.S. Bureau of Reclamation (Reclamation)’s Central Valley Project (CVP), which is operated jointly with the California Department of Water Resources’ State Water Project (SWP), manages the collection, storage, and transport of many millions of acre-feet of water through the Central Valley for delivery to irrigators and municipalities and to meet state and federal ecological and species requirements. In 2018, California finalized revisions to its Water Quality Control Plan for the San Francisco Bay and San Joaquin-Sacramento River Delta (Bay-Delta) to require that more flows from the San Joaquin and Sacramento Rivers would reach the Bay-Delta for water quality and fish and wildlife enhancement, accordingly reducing water supplies for agricultural irrigators. In 2019, the previous Trump administration responded by committing to increasing CVP water supplies for agricultural users through changes to long-term operations of the CVP, pursuant to a 2019 ESA biological opinion or “BiOp.”
These ESA changes were promptly challenged by California and environmental organizations as insufficiently protective of Bay-Delta salmon and smelt populations, habitats, and spawning activities. They were first enjoined by federal court and later remanded to the National Marine Fisheries Service (NMFS) and FWS under the Biden administration. The cases were stayed during NMFS and FWS’s reconsideration of new CVP and SWP operating rules, in favor of an interim operations plan (IOP), which was extended through December 2024 to allow for the issuance of new CVP and SWP BiOps. See March 28, 2024 Order in Pacific Coast Federation of Fishermen’s Associations v. Raimondo, Civ. Nos. 20-00426, -00431 (E.D. Cal.). On December 20, 2024, on the verge of another change in administration, Reclamation issued its Record of Decision for the “Long-Term Operation of the Central Valley Project and State Water Project” based on 2024 BiOps, to mixed reviews from environmentalists and water users alike. It is likely that these new “California water rules” will spark new rounds of both litigation challenges and regulatory reconsideration in 2025.
Yurok Tribe v. Klamath Water Users Association
In this appeal before the Ninth Circuit (Nos. 23-15499 and 23-15521, consolidated), the Klamath Water Users Association (KWA) and Klamath Irrigation District (KID) sought review of a 2023 federal district court decision holding that an Oregon Water Resources Department (OWRD) order prohibiting Reclamation from releasing stored water subject to adjudicated irrigation rights from Upper Klamath Lake to protect and restore endangered fish species was preempted by the ESA. KWA and KID had sought declaratory relief that the ESA does not authorize Reclamation to release water from Upper Klamath Lake, arguing that the case does not involve any issue of preemption, because Reclamation does not have authority under its enabling act to appropriate rights to use water in violation of Oregon law, and the ESA does not expand these Reclamation authorities. OWRD subsequently withdrew its order.
The Ninth Circuit heard oral argument on June 12, 2024, but the court, just prior to the hearing, indicated that it perceived potential jurisdictional issues due to the OWRD withdrawal having mooted the initial challenge to its order. At oral argument, KID urged the court to certify key questions to the Oregon Supreme Court concerning Reclamation’s authority to use and control the use of water under Oregon law, arguing that Oregon’s water rights and laws governing the use and control of water in Upper Klamath Lake were established long before the ESA was enacted, that Section 8 of the Reclamation Act mandates compliance with state water law and water rights, and that controlling precedent makes clear that state law governs whether Reclamation has authority or discretion to meet its ESA obligations using stored irrigation water subject to adjudicated water rights. Therefore, these state law questions should be addressed independently of the federal question of Reclamation’s ESA obligations and their preemptive consequences. Briefing on KID’s motion for certification continued into December 2024, so a Ninth Circuit ruling on the merits, or as to whether the questions will proceed for now in state or federal court, can be expected in 2025.
Water
On November 20, 2024, EPA Region 9 published in the Federal Register its Final Designation of formerly unregulated stormwater discharges from commercial, industrial, and institutional (CII) properties for required National Pollutant Discharge Elimination System (NPDES) stormwater permitting. The designation applies to CII facilities consisting of five or more acres of impermeable surfaces (in the case of unpermitted facilities) or five or more total acres (in the case of unpermitted portions of facilities already holding a NPDES permit and no exposure certificate, and in the case of non-notice of non-applicability (NONA) covered portions of facilities with a NONA) in two watersheds in the Los Angeles County area. This expansion of stormwater regulation is a joint effort between EPA Region 9 and the Los Angeles Regional Water Quality Control Board. The Water Board prepared the corresponding draft CII General Permit and is expected to hold a public hearing on the draft permit now that EPA’s designation is final.
The incoming Trump administration may reevaluate the Final Designation and consider rescinding it, but it may take some time for new EPA staffers to address this action. In the interim, it will be critical for parties adversely affected by the Final Designation to expeditiously seek judicial review—and a stay or preliminary injunction—to protect their interests.
Additional Authors: Gary J. Smith, Patrick J. Redmond, Leticia E. Duarte, Sara M. Eddy, Gabriela Espir, Jeremy D. Faulkner, Nicole L. Garson, Ragini Gupta, Lauren M. Lankenau, Sharon Mathew, Claire S. McLeod Ruiz, Lauren M. Murvihill, and Megan V. Unger
Colorado Attorney General Announces Adoption of Amendments to Colorado Privacy Act Rules + Attorneys General Oppose Clearview AI Biometric Data Privacy Settlement
Colorado Adopts Amendments to CPA Rules
The Colorado Attorney General announced the adoption of amendments to the Colorado Privacy Act (“CPA”) rules. The rules will become effective on January 30, 2025. The rules provide enhanced protections for the processing of biometric data as well as the processing of the online activities of minors. Specifically, companies must develop and implement a written biometric data policy, implement appropriate security measures regarding biometric data, provide notice of the collection and processing of biometric data, obtain employee consent for the processing of biometric data, and provide a right of access to such data. In the context of minors, the amendment requires that entities obtain consent prior to using any system design feature designed to significantly increase the use of an online service of a known minor and to update the Data Protection Assessments to address processing that presents heightened risks to minors. Entities already subject to the CPA should carefully review whether they may have heightened obligations for the processing of employee biometric data, a category of data previously exempt from the scope of the CPA.
Attorneys General Oppose Clearview AI Biometric Data Privacy Settlement
A proposed settlement in the Clearview AI Illinois Biometric Information Privacy Act (“BIPA”) litigation is facing opposition from 22 states and the District of Columbia. The Attorneys General of each state argue that the settlement, which received preliminary approval in June 2024, lacks meaningful injunctive relief and offers an unusual financial stake in Clearview AI to plaintiffs. The settlement would grant the class of consumers a 23 percent stake in Clearview AI, potentially worth $52 million, based on a September 2023 valuation. Alternatively, the class could opt for 17 percent of the company’s revenue through September 2027. The AGs contend the settlement doesn’t adequately address consumer privacy concerns and the proposed 39 percent attorney fee award is excessive. Clearview AI has filed a motion to dismiss the states’ opposition, arguing it was submitted after the deadline for objections. A judge will consider granting final approval for the settlement at a hearing scheduled on January 30, 2025.